
Abstract
In patients with multiple sclerosis (MS), a diffuse axonal degeneration occurring throughout the white matter of the central nervous system causes progressive neurologic disability. The underlying mechanism is unclear. This review describes a number of pathways by which dysfunctional astrocytes in MS might lead to axonal degeneration. White-matter astrocytes in MS show a reduced metabolism of adenosine triphosphate-generating phosphocreatine, which may impair the astrocytic sodium potassium pump and lead to a reduced sodium-dependent glutamate uptake. Astrocytes in MS white matter appear to be deficient in β2 adrenergic receptors, which are involved in stimulating glycogenolysis and suppressing inducible nitric oxide synthase (NOS2). Glutamate toxicity, reduced astrocytic glycogenolysis leading to reduced lactate and glutamine production, and enhanced nitric oxide (NO) levels may all impair axonal mitochondrial metabolism, leading to axonal degeneration. In addition, glutamate-mediated oligodendrocyte damage and impaired myelination caused by a decreased production of
The majority of patients with multiple sclerosis (MS) begin with a relapsing-remitting course, which often after several years of disease duration converts into a progressive disease (secondary progressive MS). In a minority of patients, progressive neurologic deterioration without remission occurs from the disease onset (primary progressive MS). Multiple sclerosis is traditionally viewed as a T cell-driven autoimmune disease against myelin of the central nervous system (Compston and Coles, 2008). Substantial evidence indicates that inflammation has a key role in the development of focal demyelinating lesions that constitute the pathological substrate for relapses. However, the progressive phase of MS reflects a poorly understood insidious axonal degeneration that is age related and independent of relapses (Confavreux and Vukusic, 2006; Koch et al, 2007a). Pathological studies have shown that axonal degeneration occurs diffusely throughout the normal appearing white matter (Evangelou et al, 2000). This neurodegenerative component is associated with inflammation (Frischer et al, 2009), but there is growing awareness that inflammatory mechanisms alone cannot explain this degenerative process. Immunomodulatory drugs, which reduce the development of focal lesions and relapses, are not effective in progressive MS (Wilkins and Scolding, 2008), and pathological studies show ongoing demyelination and axonal degeneration despite pronounced immunosuppression (Metz et al, 2007).
Axonal degeneration might be caused by Wallerian degeneration secondary to axonal injury in focal demyelinating lesions (Ferguson et al, 1997; Trapp et al, 1998). However, both magnetic resonance imaging and neuropathological studies found a lack of correlation between focal lesion load and axonal loss in the spinal cord (Bergers et al, 2002; DeLuca et al, 2006; Kutzelnigg et al, 2005), indicating that mechanisms other than Wallerian degeneration contribute to this progressive axonal degeneration.
A current hypothesis suggests that axonal mitochondrial energy failure may lead to axonal degeneration in MS (Su et al, 2009; Trapp and Stys, 2009). There is an increasing awareness that astrocytes have an important role in a variety of neurodegenerative diseases (De Keyser et al, 2008; Kimelberg and Nedergaard, 2010; Ransom et al, 2003), but their possible role in the pathogenesis of MS has received little attention. This review describes a number of pathways by which dysfunctional white-matter astrocytes in MS might lead to axonal mitochondrial energy failure and axonal degeneration.
Phosphocreatine metabolism
Phosphorus magnetic resonance spectroscopy of the brain produces multiple peaks representing high-energy phosphorus compounds, including phosphocreatine (PCr) and adenosine triphosphate (ATP). Minderhoud et al (1992) found that compared with healthy controls, MS patients had increased PCr/β-ATP ratios in the centrum semiovale, and this correlated with clinical measures of MS severity. The β-ATP peak does not contain contributions from other components and appears to be constant under different metabolic conditions (van der Knaap and Pouwels, 2005), suggesting that PCr levels were elevated. Husted et al (1994) found significantly increased PCr/total 31P ratio values in the normal appearing white matter of the centrum semiovale, but not in focal MS lesions of MS patients versus healthy controls. Steen et al (2010) corroborated these findings by detecting significantly increased PCr/β-ATP and PCr/total 31P ratios in the normal appearing white matter of the centrum semiovale of MS patients, compared with healthy controls.
The results from these three independent studies indicate that PCr levels in the normal appearing white matter of MS patients are increased, suggesting that this source of energy generated by mitochondrial creatine kinase (CK) is not properly used.
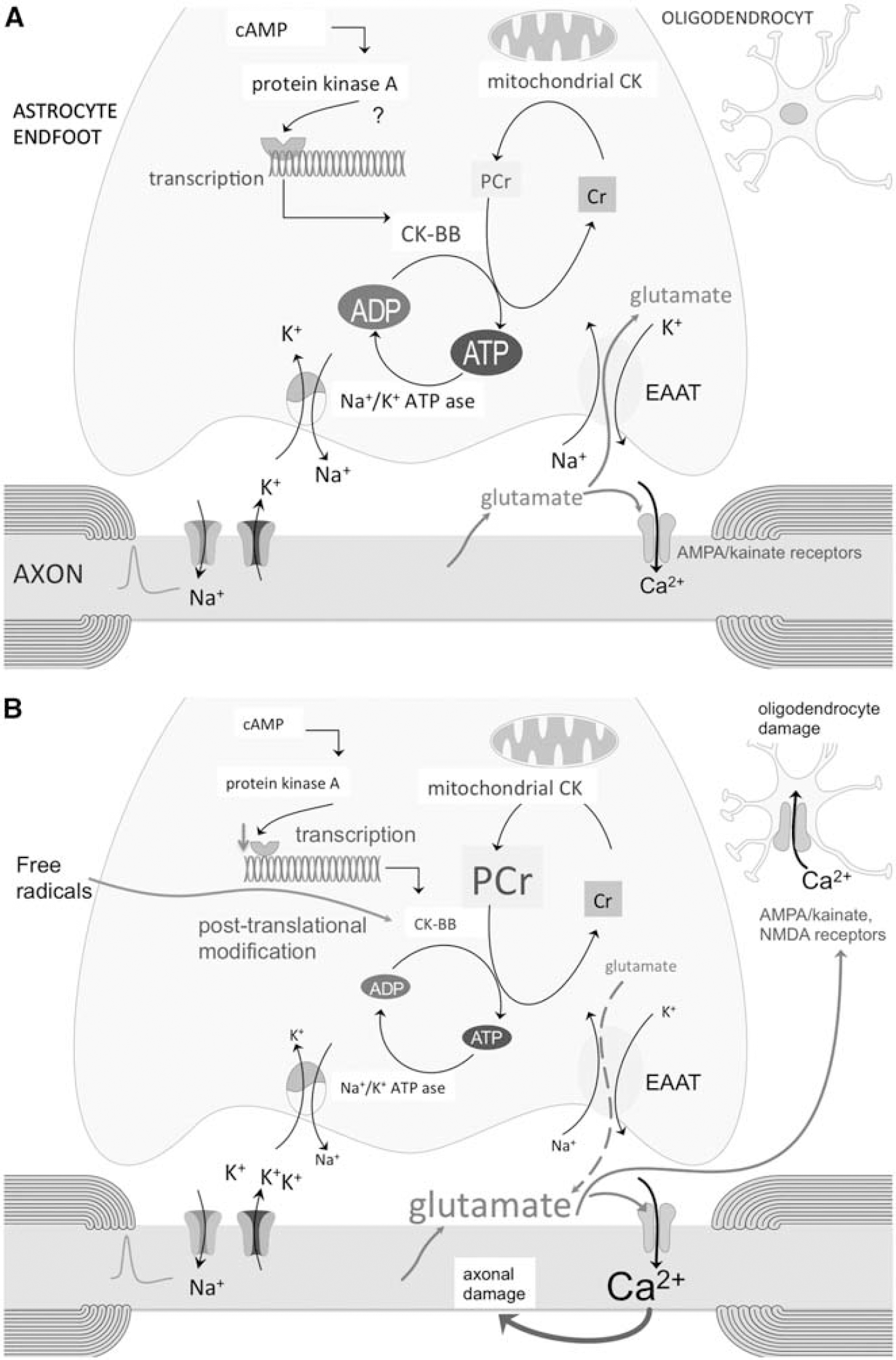
Phosphocreatine acts as a metabolic buffer that is transported from mitochondria to high-energy consuming areas in the cytosol (Figure 1A). Cytosolic CK catalyzes the reversible transfer of the phosphor group from PCr to ADP, generating ATP at a much faster rate than glycolysis and oxidative phosphorylation (Brosnan and Brosnan, 2007). The brain cytosolic CK isoform is CK-BB, which in white matter appears to be exclusively present in astrocytes in both rat and human brain (Tachikawa et al, 2004; Thompson et al, 1980).
(
Mechanisms that may lead to a decreased CK-BB concentration in cerebral white matter are a reduced transcription or posttranslational modification of the enzyme. Decreased transcription might be caused by a deficiency of astrocytic β2 adrenergic receptors in MS white matter (De Keyser et al, 1999; Zeinstra et al, 2000). Activation of these receptors by norepinephrine increases the levels of intracellular cAMP. Norepinephrine, activating these β2 adrenergic receptors, is probably released from axonal varicosities that are present along noradrenergic axons throughout the white matter (Chiti and Teschemacher, 2007). Reduction of intracellular cAMP levels impairs the transcription of CK-BB in cultured human U87-MG glioblastoma cells (Kuzhikandathil and Molloy, 1994, 1999).
The mechanism underlying a loss of β2 adrenergic receptors on astrocytes in MS is unclear. The same finding has been made in dogs following an encephalomyelitis caused by the canine distemper morbillivirus. This condition leads to a chronic demyelinating disease that is very similar to MS, including an axonal degeneration throughout the normal appearing white matter (Seehusen and Baumgartner, 2010; Vandevelde and Zurbriggen, 2005). Although an infectious component has long been suspected, no specific transmissible agent has so far been linked convincingly to MS. Searches for the presence of a morbillivirus in postmortem MS brain tissue have been inconclusive (Geeraedts et al, 2004; Lassmann et al, 2003).
Posttranslational modification of CK-BB might be caused by free radicals, especially by the oxidation of thiol groups of its structure (Wolosker et al, 1996). There are indications that posttranslational oxidative modification of the enzyme may contribute to decreased CK-BB activity in the cerebral cortex in a number of neurodegenerative disorders, including Alzheimer's disease (Aksenov et al, 2000; Aksenov et al, 1999). In MS, there is increased production of reactive oxygen species, not only in focal inflammatory white-matter lesions (Langemann et al, 1992), but also throughout the normal appearing white matter (Graumann et al, 2003). Further research is needed to find out which mechanism is primarily involved.
Possible consequences of an impaired phosphocreatine metabolism
The most ATP consuming activity in astrocytic end feet during axonal electrogenesis is the Na+/K+-ATP pump. It takes up K+ released by axons in the extracellular space after each depolarization, and it establishes the Na+ gradient necessary for glutamate uptake by the astrocytic Na+-dependent glutamate transporters (Figure 1A; Anderson and Swanson, 2000; Danbolt, 2001). Axonal release of glutamate is thought to represent a widespread mechanism for activity-dependent signaling at the axon—glia interface throughout the white matter. Glutamate in white-matter axons is stored in vesicles, and the propagation of action potentials along these axons leads to rapid vesicular release of glutamate into the extracellular fluid by exocytosis (Kukley et al, 2007; Ziskin et al, 2007). White-matter axons contain voltage-gated Ca2+ channels and the vesicle fusion machinery that are necessary for this type of activity-mediated exocytosis (Alix and Domingues, 2011). The functional significance of this vesicular glutamate release is unclear. After stimulation of white-matter axons, synaptic-like potentials mediated by α-amino-3-hydroxyl-5-methyl-4-isoxazole-propionic acid (AMPA) receptors have been recorded in patch-clamped NG2+ glia (Kukley et al, 2007).
In human white matter, the Excitatory Amino Acid Transporter 1 is expressed in oligodendrocytes and astrocytes, whereas Excitatory Amino Acid Transporter 2, which has the largest role in regulating extracellular glutamate concentration, is essentially located throughout the processes of astrocytes (Domercq and Matute, 1999; Vallejo-Illarramendi et al, 2006). These transporters move glutamate into astrocytes and oligodendrocytes against a steep concentration gradient by coupling glutamate translocation to the transmembrane Na+, K+ gradients. These gradients are maintained by the membrane Na+/K+-ATP pump, such that glutamate uptake is ultimately ATP dependent.
Reduced activity of the astrocytic Na+/K+-ATP pump will lead to high extracellular K+ concentrations and reversal of glutamate uptake by glutamate transporters (Figure 1B; Rose et al, 2009). Peripheral astrocytic processes containing mitochondria (Lovatt et al, 2007) terminate as fine astrocytic endings that contact the axonal nodes (Raine, 1984). There is evidence from
Compared with healthy controls, subjects with MS have increased glutamate levels throughout the normal appearing white matter (Srinivasan et al, 2005), and in the cerebrospinal fluid (Sarchielli et al, 2003). Central myelinated axons express functional kainate and Ca2+ permeable AMPA receptors, which on overstimulation by glutamate may lead to excitotoxic damage of axons, caused by an increased influx of Ca2+(Ouardouz et al, 2009a, 2009b).
Oligodendrocytes, expressing AMPA/kainate and
Axonal energy metabolism
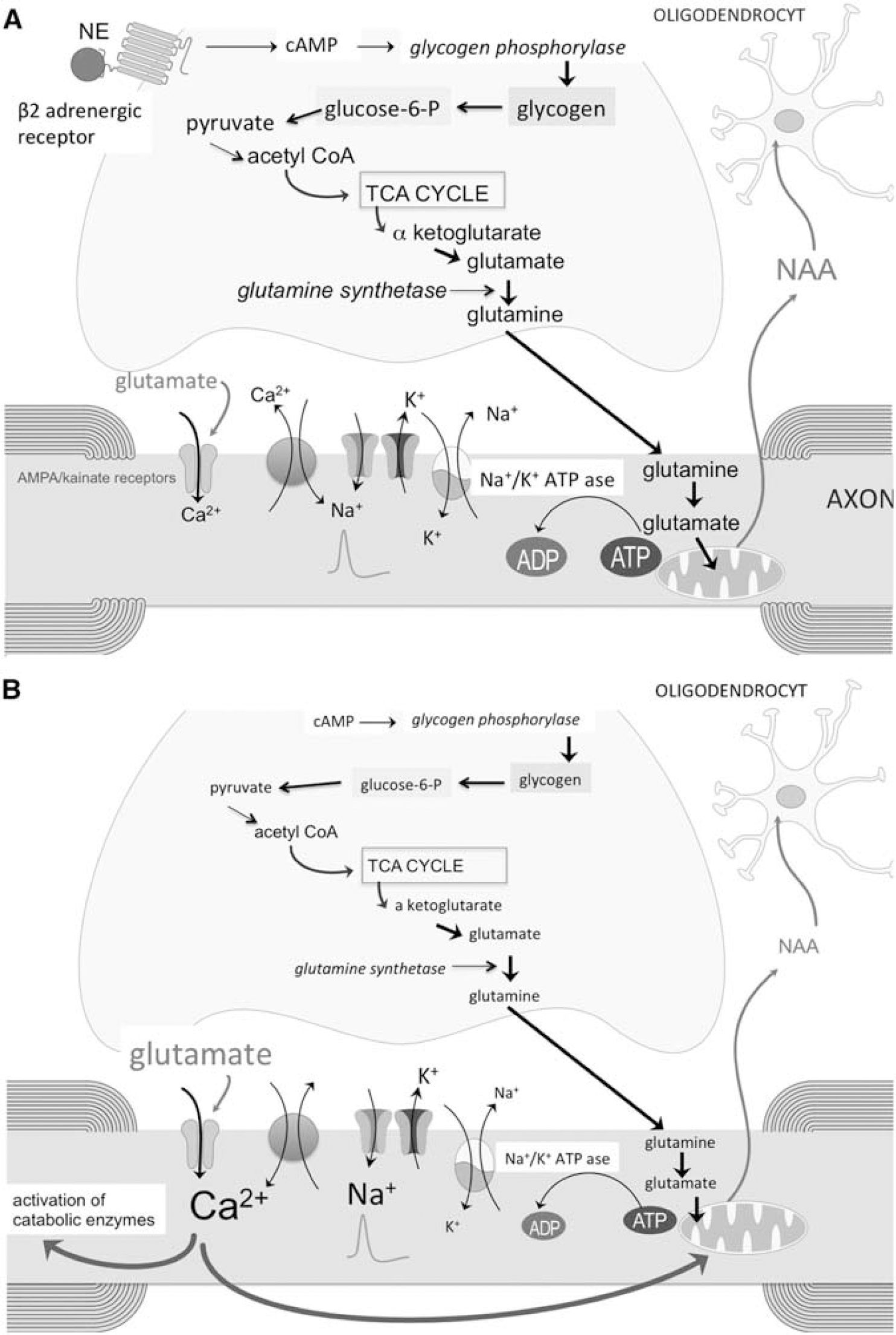
Axonal mitochondrial ATP metabolism and the synthesis of NAA are indirectly linked (Figure 2). Aspartate aminotransferase facilitates the conversion of glutamate to α-ketoglutarate and oxaloacetate to aspartate. Glutamate in neurons and their axons is mainly derived from glutamine that is shuttled from astrocytes to neurons, because pyruvate carboxylase is a glia-specific enzyme (Hertz et al, 2007). Pyruvate kinase converts pyruvate to oxaloacetate. After condensation of oxaloacetate with acetyl CoA, the resulting citrate molecule can be converted to α-ketoglutarate and subsequently to glutamine. There are some indications that carboxylation of pyruvate, supporting the formation of glutamate, may also occur in neurons, but the mechanism is not clear (Fan et al, 2010; Hassel, 2001). Acetylation of aspartate by the neuronal enzyme aspartate
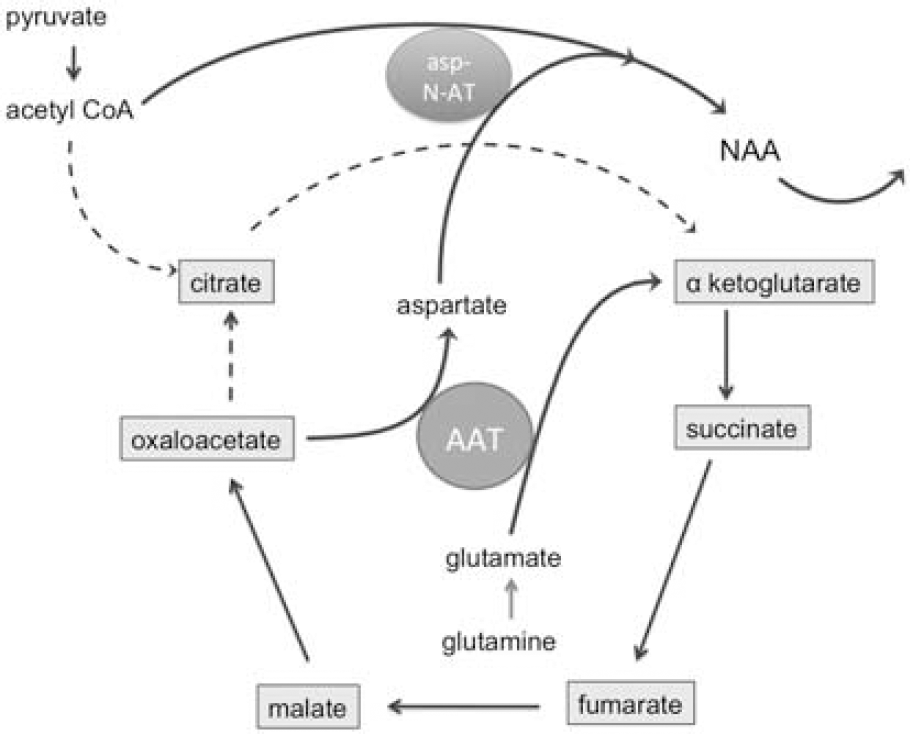
Schematic representation of the possible mechanism underlying
Several 1H magnetic resonance spectroscopy studies of the normal appearing white matter in MS subjects showed decreased levels of NAA compared with healthy controls (Aboul-Enein et al, 2010; Chard et al, 2002; De Stefano et al, 1998, 2001, 2002; Fu et al, 1998; Leary et al, 1999; Lee et al, 2000). Decreased NAA levels in normal appearing white matter are already present at the early stages of the disease and progresses over time (De Stefano et al, 2001). Cader et al (2007) investigated the relationship between callosal size, diffusion magnetic resonance imaging parameters, and NAA concentrations in the corpus callosum in MS patients. Relative changes in NAA were not directly related to diffusion-derived parameters of axonal loss. Another study in MS patients estimated the structural contributions to NAA, as assessed by axial diffusivity derived from diffusion tensor imaging and cross-sectional volumetric imaging in the spinal cord (Ciccarelli et al, 2010b). Lower residual variance in NAA, reflecting information specific to axonal mitochondrial metabolism, was associated with greater clinical disability independent of structural damage. These findings support the idea that metabolic mitochondrial dysfunction in axons, and not just axonal loss, is an important determinant of reduced NAA concentrations in the normal appearing white matter of MS patients.
Reductions of NAA in normal appearing white matter of drug naive MS subjects were partially reversible in a 2-year longitudinal assessment in the early stages of relapsing-remitting MS (Tiberio et al, 2006). Only 30% of the patients in this study started with interferon β during the evaluation period. Spontaneous recovery of NAA concentrations in focal lesion has also been documented in brain and spinal cord of MS patients (Ciccarelli et al, 2010a; Davie et al, 1994). These spontaneous improvements may reflect either restored mitochondrial activity or a compensatory increase in the number of mitochondria. Histopathological studies found increased numbers of mitochondria and an upregulation of mitochondrial cytochrome C oxidase (complex IV) in axons in both chronic lesions and normal appearing white matter of MS subjects (Mahad et al, 2009; Witte et al, 2009).
An increase in NAA levels in normal appearing white matter of relapsing-remitting MS subjects has been reported after 2 years of treatment with glatiramer acetate (Khan et al, 2005), and after 1 year of treatment with β-interferon (Narayanan et al, 2001). However, numbers of patients in these studies were low, and spontaneous improvement as observed in early onset MS could not be excluded. Another study found no effect of 1-year treatment with β-interferon on white-matter NAA levels in relapsing-remitting MS (Parry et al, 2003). More targeted therapies to improve axonal mitochondrial function should be able to show energy recovering effects within days or weeks. The oral administration of fluoxetine, which is believed to stimulate glycogenolysis in astrocytes, significantly increased the NAA/Cr ratio in cerebral white matter of MS patients within 2 weeks (Mostert et al, 2006).
Possible causes of reduced axonal mitochondrial function
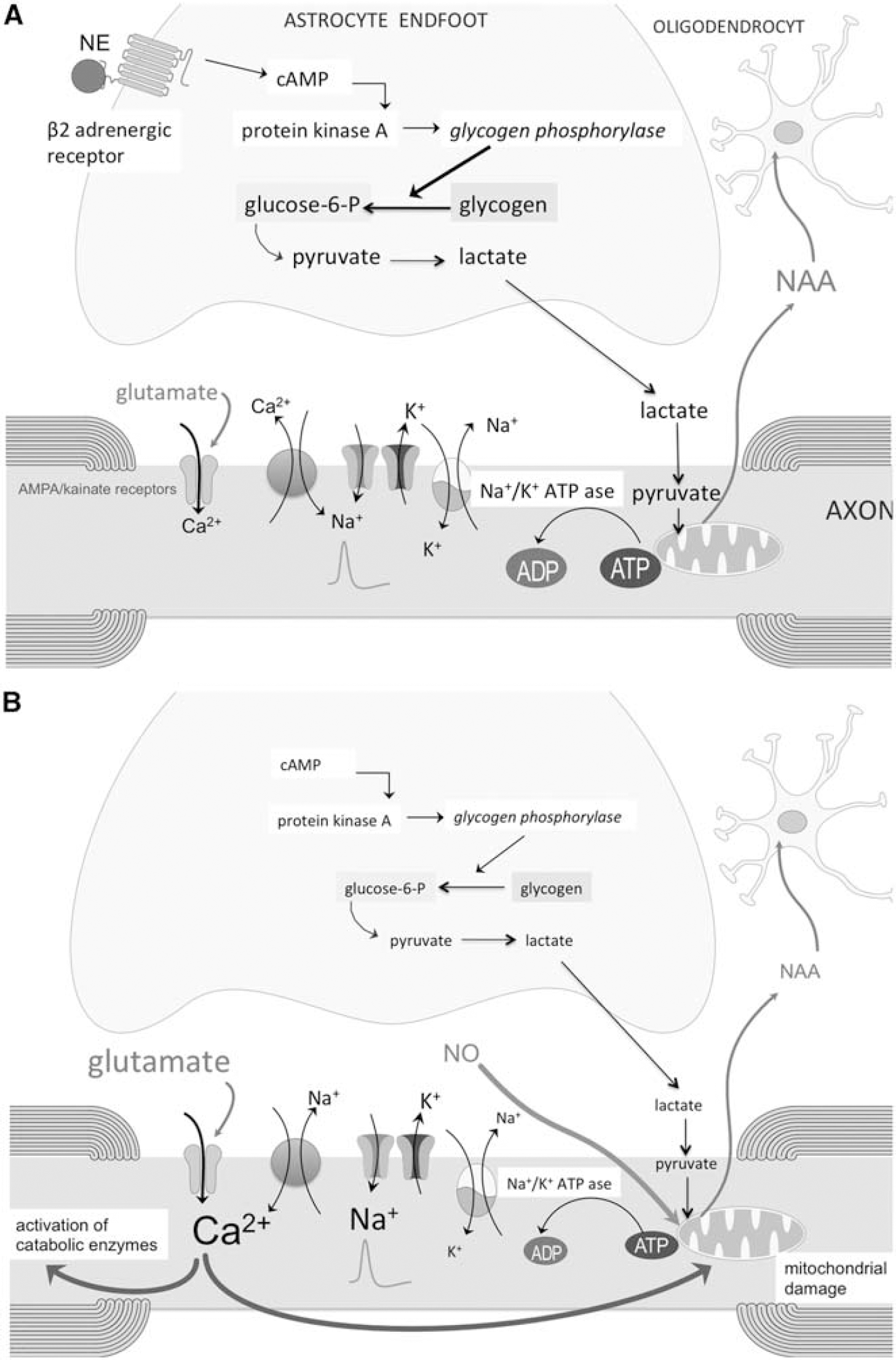
A number of mechanisms may lead to a reduced axonal mitochondrial energy metabolism in the normal appearing white matter of subjects with MS (Figures 3 and 4). A first mechanism may be related to nitric oxide (NO). Nitric oxide synthase (NOS2) is increased in both active focal lesions and throughout the normal appearing white matter (Broholm et al, 2004). On immunostaining, NOS2-positive cells appear to be predominantly astrocytes, and norepinephrine or other agents that elevate cAMP inhibit the expression of NOS2 in astrocytes (Feinstein, 1998; Feinstein et al, 1993). A loss of astrocytic β2 adrenergic receptors in MS might explain why astrocytes in MS plaques and normal appearing white matter express high levels of NOS2 (De Keyser et al, 2004). Enhanced levels of NO can compete with oxygen for the binding domain on cytochrome C oxidase (complex IV in the mitochondrial respiratory chain). This may reduce the electron flow and subsequently ATP synthesis. The increased expression and activity of cytochrome C oxidase, observed in chronic lesions and normal appearing white matter of MS subjects, might represent a compensation mechanism to overcome NO occupancy of the enzyme (Mahad et al, 2009; Witte et al, 2009).
(
(
A second mechanism is toxicity caused by increased glutamate levels, which have been detected throughout the normal appearing white matter (Srinivasan et al, 2005). Enhanced extracellular glutamate levels might result from a reduced astrocyte energy metabolism and failure of glutamate uptake (see above). An increased influx of Ca2+ into axons by overstimulation of AMPA/kainate receptors (Ouardouz et al, 2009a, 2009b) may damage mitochondria by promoting Ca2+ entry into the matrix, opening of the permeability transition pore, and release of cytochrome C into the cytosol (Gunter et al, 2004).
A third possible mechanism is an impaired glycogenolysis caused by a deficiency of β2 adrenergic receptors in white-matter astrocytes (De Keyser et al, 1999; Zeinstra et al, 2000). Astrocytes are the main glycogen reservoir in the central nervous system.
A fourth mechanism to be considered is a reduced astrocytic oxidative metabolism and decreased astrocytic synthesis of glutamine. This would impair the glutamine shuttle as energy source for axons (Figures 2, 4A, and 4B; Hertz and Gibbs, 2009). Compared with controls, several nuclear-encoded mitochondrial genes and the functional activities of mitochondrial respiratory chain complexes I and III were decreased in motor cortex from MS patients (Dutta et al, 2006). The authors claimed that the reduced mitochondrial gene expression was specific for neurons, but this is difficult to prove since protoplasmic astrocytes in motor cortex also exhibit robust oxidative metabolism (Lovatt et al, 2007). Whether a reduced oxidative metabolism occurs in white-matter astrocytes in MS has not been studied. Compared with myelinated hippocampus and demyelinated motor cortex, demyelinated hippocampus dissected from postmortem MS brains showed a downregulation of glutamine synthetase (Dutta et al, 2011). A deficiency in white-matter astrocytic β2 adrenergic receptors and reduced glycogenolysis in MS might impair astrocytic oxidative metabolism and glutamine synthetase activity. Support for this hypothesis comes from studies showing that the addition of dibutyryl-cAMP, which is a cell permeable cAMP analog, enhanced glutamine synthetase activity in astrocyte cultures (Brookes, 1992; Stanimirovic et al, 1999).
Enhanced consumption of ATP leads to an increase in oxypurines (uric acid, hypoxanthine, and xanthine) and purine nucleosides (inosine, adenosine, and guanosine), which are ATP breakdown products. Higher cerebrospinal fluid and serum concentrations of these end products were found in MS subjects compared with controls (Amorini et al, 2009; Lazzarino et al, 2010). In a follow-up study, higher baseline ATP metabolites were associated with a more severe progression of disability and brain atrophy 3 years later, suggesting that an increased energy demand precedes the axonal degeneration in MS. Neuron-specific enolase is a critical enzyme in neuro-axonal glycolysis, where it converts 2-phospho-D glycerate to phosphoenolpyruvate. Multiple sclerosis patients with a clinically relevant progression of disability after 5 years of follow-up had lower baseline plasma neuron-specific enolase levels than those who remained clinically stable (Koch et al, 2007b), supporting the hypothesis that reduced axonal metabolic activity may precede axonal degeneration and progression of disability in MS.
Consequences of axonal energy failure
A decreased ATP production by mitochondria reduces the activity of the axolemmal Na+/K+ pump, leading to intraaxonal accumulation of Na+ and relative axonal depolarization. This may open voltage-gated Ca2+ channels and induce reversal of the Na+/K+ exchanger, leading to intraaxonal accumulation of Ca2+ (Figure 3). The intraaxonal Ca2+ overload will damage mitochondria (see above) and inappropriately stimulate a variety of Ca2+-dependent catabolic enzyme systems, including proteases, phospholipases, and calpains, ultimately leading to axonal degeneration (Stys, 2005). In addition, reduced formation of NAA may impair myelin membrane turnover and lead to loss of myelin. The maintenance of myelin requires a continuous and dynamic process of myelin component catabolism and recycling (Ando et al, 2003).
Benign multiple sclerosis
It has long been recognized that a subgroup of individuals with MS shows little or no progression in severity of the disease over time. This so-called “benign MS” can be arbitrarily defined by minimal or no disability after at least 10 years of observation (Ramsaransing and De Keyser, 2006). A less axonal degenerative process appears to be present in this subset of MS patients (Gauthier et al, 2009). Compared with controls, patients with benign MS only showed a nonsignificant trend for elevated PCr ratios in the normal appearing white matter (Steen et al, 2010), and NAA levels in the normal appearing white matter were also relatively preserved (Benedetti et al, 2009; Davie et al, 1997; Steen et al, 2010). Patients with a relatively benign course of MS therefore represent an interesting subgroup to better understand compensatory mechanisms of astrocytic and axonal energy metabolism. It is not known whether patients with benign MS have less elevated glutamate levels in the normal appearing white matter. Genetic association studies may give worthwhile clues. A recent genome-wide association analysis in patients with MS found a significant association between genes with high relevance to glutamate biology and both brain glutamate levels and the degree of neurodegeneration over 1 year of follow-up (Baranzini et al, 2010). This finding needs to be confirmed, and the role of the gene products clarified.
Conclusion
Evidence is evolving that a defective axonal energy metabolism has a role in the diffuse axonal degeneration in MS. A number of findings suggest that at least part of this defective axonal metabolism might be secondary to astrocyte dysfunction. A deficiency in astrocytic β2 adrenergic receptors may be responsible for a reduced glycogenolysis, resulting in a decreased formation of lactate and glutamine, which are energy sources for axons, and for increased levels of NO. An impaired PCr metabolism in astrocytes may lead to increased levels of glutamate in the extracellular space surrounding axons. All these mechanisms can converge to an intraaxonal Ca2+ overload that further impairs mitochondrial function and stimulates catabolic enzymes, leading to axonal degeneration. In addition, decreased NAA levels and excitotoxic damage of oligodendrocytes may impair axonal myelination, and contribute to axonal degeneration. A better insight into these different processes and the protective factors that underlie a relatively benign disease course might ultimately lead to new therapies that slow down the progression of disability in patients with MS.
Footnotes
Acknowledgements
Our research on the role of astrocytes in MS is supported by FWO Belgium (http://www.fwo.be) and the Belgian Charcot Foundation (![]() ).
).
The authors declare no conflict of interest.