
Abstract
Since its introduction 16 years ago, the astrocyte–neuron lactate shuttle (ANLS) model has profoundly modified our understanding of neuroenergetics by bringing a cellular and molecular resolution. Praised or disputed, the concept has never ceased to attract attention, leading to critical advances and unexpected insights. Here, we summarize recent experimental evidence further supporting the main tenets of the model. Thus, evidence for distinct metabolic phenotypes between neurons (mainly oxidative) and astrocytes (mainly glycolytic) have been provided by genomics and classical metabolic approaches. Moreover, it has become clear that astrocytes act as a syncytium to distribute energy substrates such as lactate to active neurones. Glycogen, the main energy reserve located in astrocytes, is used as a lactate source to sustain glutamatergic neurotransmission and synaptic plasticity. Lactate is also emerging as a neuroprotective agent as well as a key signal to regulate blood flow. Characterization of monocarboxylate transporter regulation indicates a possible involvement in synaptic plasticity and memory. Finally, several modeling studies captured the implications of such findings for many brain functions. The ANLS model now represents a useful, experimentally based framework to better understand the coupling between neuronal activity and energetics as it relates to neuronal plasticity, neurodegeneration, and functional brain imaging.
Introduction
Over 60 years of clinical and experimental studies of brain energy metabolism in a variety of species including humans have yielded the unquestioned evidence that, under normal physiological conditions in the adult brain, glucose is the almost exclusive energy substrate and that it is fully oxidized, resulting in a respiratory quotient close to 1 (Magistretti, 2008). Some refinements in the overall energy budget of the brain have been provided, showing for example that glucose utilization is in fact higher than that predicted by oxygen consumption in an organ with a respiratory quotient of 1. Based on stoichiometric relationships, to fully oxidize 1 mmol of the 6-carbon glucose molecule, 6 mmol of oxygen would be required. Based on oxygen consumption measurements, glucose utilization by the brain should be around 26.6 mmol/100 g of brain weight/min. Yet, glucose utilization is significantly higher in the order of 31 mmol/100 g of brain weight/min (Kety and Schmidt, 1948). The question then arises of the fate of the additional 4.4 mmol/100 g of brain weight/min. The bioenergetic reply to this question would be glycolysis, namely nonoxidative glucose utilization. If this were the case, a negative arterio-venous (A-V) difference for lactate should be measured, indicating lactate production by the brain. However, a large set of data indicate that such lactate production is marginal (Magistretti, 2008). In addition to glycolysis, and two additional metabolic pathways for glucose processing such as the pentose phosphate pathway and glycogen synthesis, other possible fates for glucose consumed by the brain in excess of oxygen consumption, may be structural, rather than energy-producing ones. Thus, glucose is a substrate for the synthesis of lipids and for amino acids, the building blocks of proteins, as well as for neurotransmitters such as glutamate, GABA (γ-aminobutyric acid), and acetylcholine. A physiological process that entails high structural demands is neuronal plasticity, as expansions of the lipid bilayer of membranes at synaptic sites and new protein synthesis are associated with such processes.
Studies at the organ level do not allow the appreciation of one of the fundamental features of brain energy metabolism, namely the fact that it is tightly coupled to synaptic activity and hence subject to spatially and temporally defined regulatory mechanisms. A major advance in the analysis of local brain glucose metabolism has been provided by Sokoloff (1981) in the 1970s with the development of the 2-deoxyglucose (2-DG) autoradiographic technique for laboratory animals, which was later applied to human with 18F-2-DG positron emission tomography (PET). While of invaluable usefulness for functional brain imaging, the spatial resolution of the 2-DG technique could not afford the identification of the cellular elements into which glucose was being predominantly taken up nor its metabolic fate after phosphorylation to 2-DG-6-phosphate. Indications that the predominant site of 2-DG uptake was in the neuropil, possibly at synaptic or perisynaptic sites, rather than the cell body of neurons, was provided by a series of elegant experiments performed by Sokoloff's laboratory (Kadekaro et al, 1987). In a striking example, providing a spatial dissociation between neuronal cell bodies and the neuropil, it was shown that stimulation of primary somatosensory fibers increased the 2-DG signal in the posterior horn of the relevant spinal cord segment, with minimal uptake into the dorsal root ganglion, where the cell bodies are localized (Kadekaro et al, 1985).
It is against this background that in the early 1990s we started a series of experiments aiming at providing a cellular resolution to the 2-DG technique and at identifying the neuronal signals that could trigger the local increases in glucose utilization associated with synaptic activity. To address these questions, two conditions were initially required: first, to have access to purified preparations of the two main cell types of the brain, namely neurons and astrocytes. Second, to test the effects of neurotransmitters released during synaptic activity on 2-DG uptake in these purified preparations. The results of this study were published in 1994 in a Proceedings of the National Academy of Sciences of the United States of America (PNAS) article entitled: ‘Glutamate uptake into astrocytes stimulates aerobic glycolysis: a mechanism coupling neuronal activity to glucose utilization’ (Pellerin and Magistretti, 1994). This is the
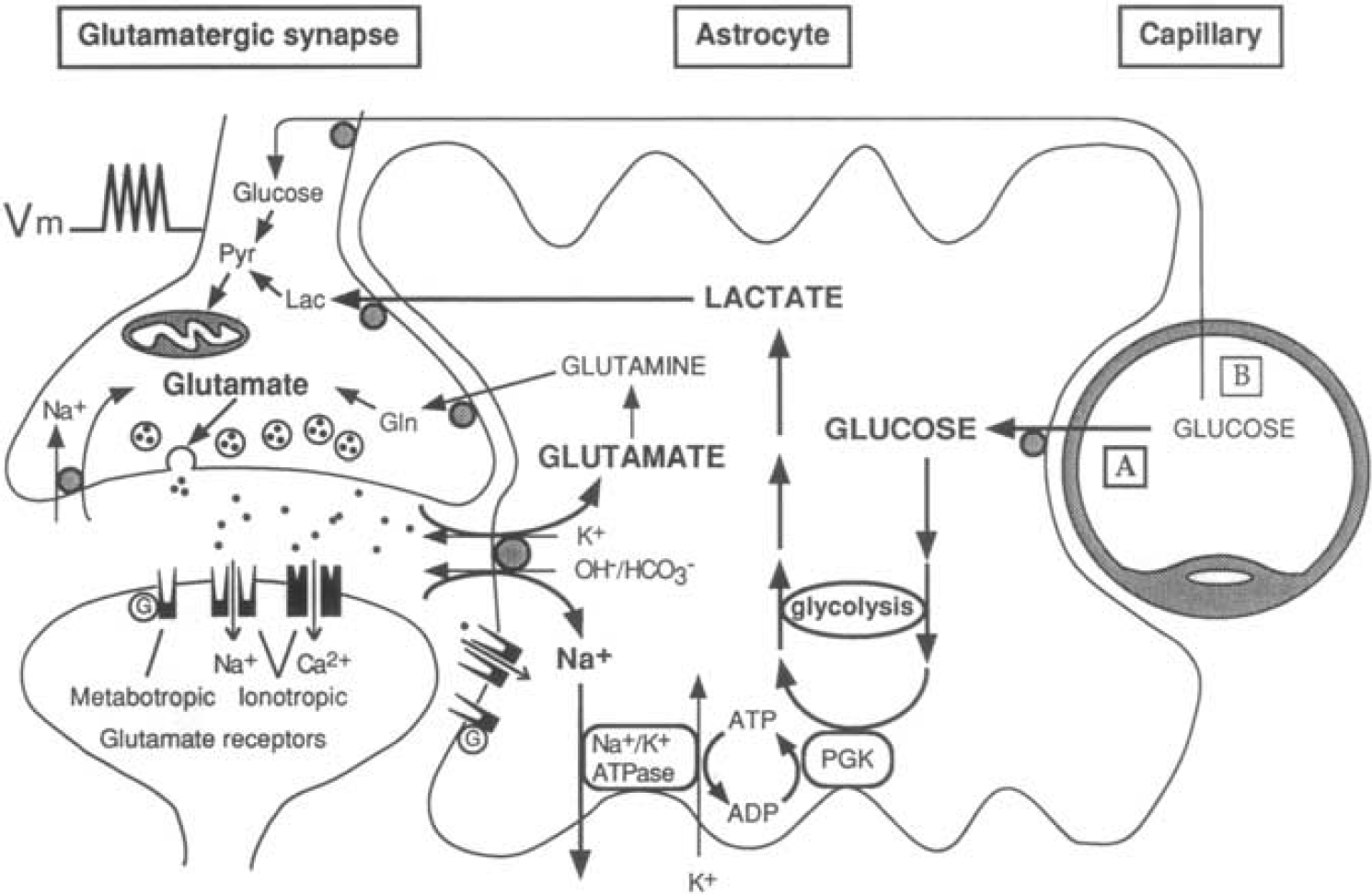
The first picture of the astrocyte–neuron lactate shuttle (ANLS) model at birth on 25 October 1994. Schematic of the mechanism for glutamate-induced glycolysis in astrocytes during physiological activation. At glutamatergic synapses, glutamate depolarizes neurons by acting at specific receptor subtypes. The action of glutamate is terminated by an efficient glutamate uptake system located primarily in astrocytes. Glutamate is cotransported with Na+, resulting in an increase in [Na+]
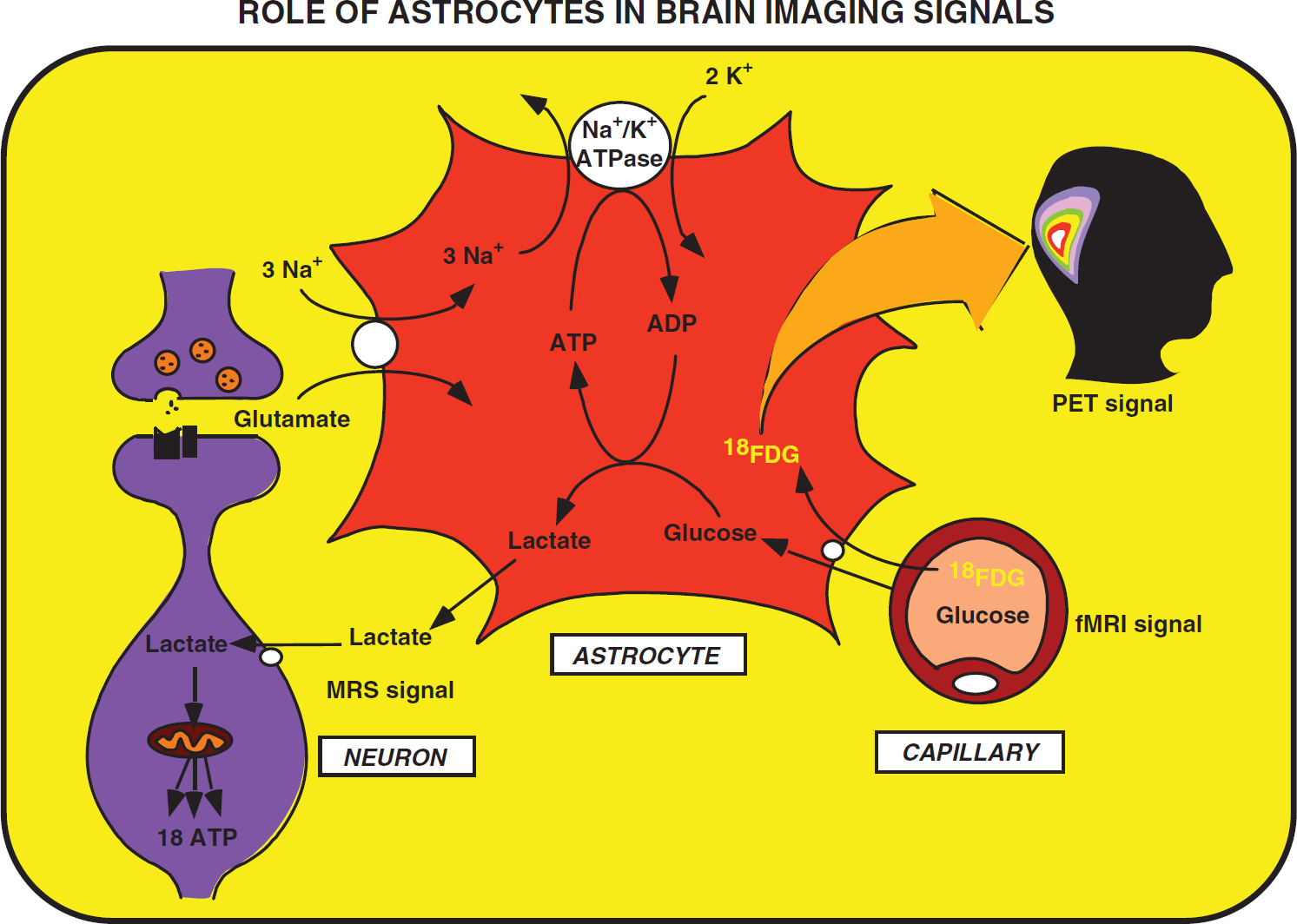
Proposed concept derived from the astrocyte–neuron lactate shuttle (ANLS) model that brain imaging signals based on glucose uptake primarily reflect astrocyte metabolism. During synaptic activity, glutamate is released into the synaptic cleft. A glutamate transporter system on astrocytes ensures removal of glutamate from the synaptic cleft. The entry of Na+ cotransported with glutamate activates the Na+/K+ ATPase. Activation of the Na+/K+ ATPase is coupled with an increased glycolytic flux, hence resulting in the stimulation of glucose uptake from the capillaries. Lactate, the major end product of glycolysis, is released by astrocytes and taken up by neurons where it can enter the tricarboxylic acid (TCA) cycle and provide 18 ATP per molecule. This model implies that the activity-linked uptake of 18Fluorodeoxyglucose (18FDG) monitored with positron emission tomography (PET) reflects primarily an astrocyte-based signal. However, since neuronally released glutamate triggers the cascade of events that leads to glucose uptake, the 18FDG-PET signal faithfully reflects activation of neuronal circuits. Taken from Magistretti and Pellerin (1996). fMRI, functional magnetic resonance imaging.
The past 16 years have seen an accumulation of experimental demonstrations that imposed the ANLS as a coherent model capable to integrate a vast array of
Astrocytes and Neurons Exhibit Distinct Cytoarchitectural and Metabolic Features Consistent with Astrocyte–Neuron Lactate Shuttle
Nineteenth century's anatomists already recognized the particular cytoarchitectural relationships entertained by astrocytes with various parenchymal elements including neurons and blood vessels. Thus, Camillo Golgi and others suggested over a 100 years ago that astrocytes are likely to be involved in the transit of energy substrates from the circulation to supply neurons (Andriezen, 1893). Indeed, astrocytes send processes forming at their extremities specialized structures called end-feet abutting on cerebral blood vessels (Kacem et al, 1998). The presence of specific glucose transporters (GLUT1) at the surface of these structures (Morgello et al, 1995; Yu and Ding, 1998) argues in favor of the idea that it represents a privileged uptake site for glucose, as it leaves the circulation to enter the brain parenchyma. In parallel, other astrocytic processes ensheath synapses, providing an interface of communications between neurons and astrocytes. Such features provide the anatomical basis to implicate astrocytes in a coupling mechanism between synaptic activity and glucose utilization as we suggested in the original description of the model (Pellerin and Magistretti, 1994). A similar parallel was recently made to support the newly described role of astrocytes in coupling synaptic activity with the local regulation of blood flow (Gordon et al, 2008), although the mechanisms for neurovascular coupling appear to be quite complex and redundant, involving also, for example, the activity of GABAergic interneurons (Kocharyan et al, 2008). Quite early on, it became clear that a single astrocyte might not be the most adequate entity to host the entire metabolic coupling mechanism. Since astrocytes are not isolated but form a syncytium through their connections via gap junctions (Giaume et al, 2010), it was proposed that energy substrates produced in response to synaptic activity would transit through several astrocytes before being delivered to stimulated neurons (Magistretti et al, 1995) (Figure 3). As predicted, evidence was recently provided demonstrating that astrocytic coupling via gap junctions contributes to the energetic support of active neurons through supply of glucose-derived lactate (Rouach et al, 2008).
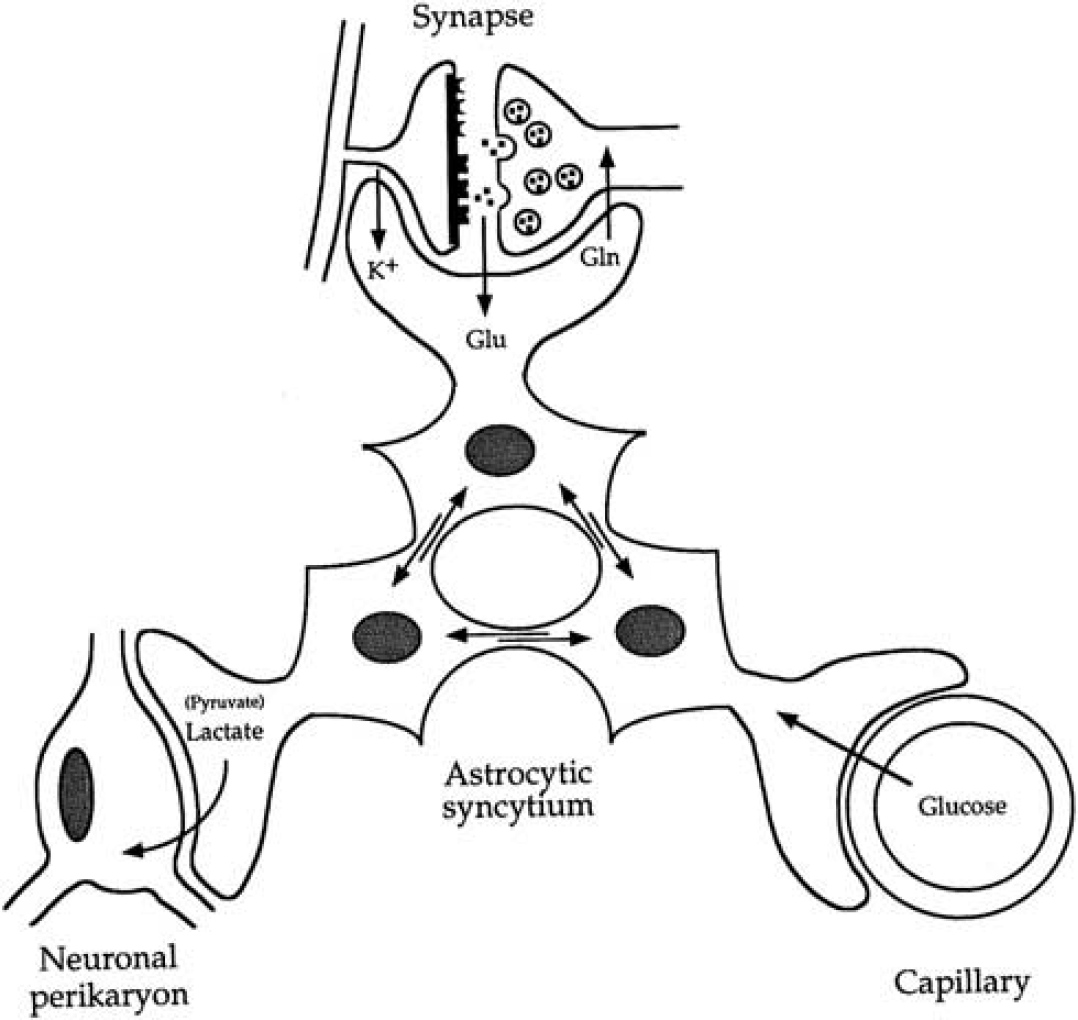
Introduction of the concept of the importance of the astrocytic syncytium for intracellular metabolite trafficking, also known as metabolic polarity of the astrocytic syncytium. Astrocytes have various processes that make contact either with capillaries, neuronal perikarya, or synapses. In addition, they are connected with each other at gap junctions; through these junctions small molecules (MW<1,000) can be exchanged. These properties endow the astrocytic syncytium with the capacity to ensure the transfer of metabolic intermediates from areas of production to areas of demand. Gln, glutamine; Glu, glutamate. Taken from Magistretti et al (1995).
Several studies have documented clear metabolic differences between astrocytes and neurons
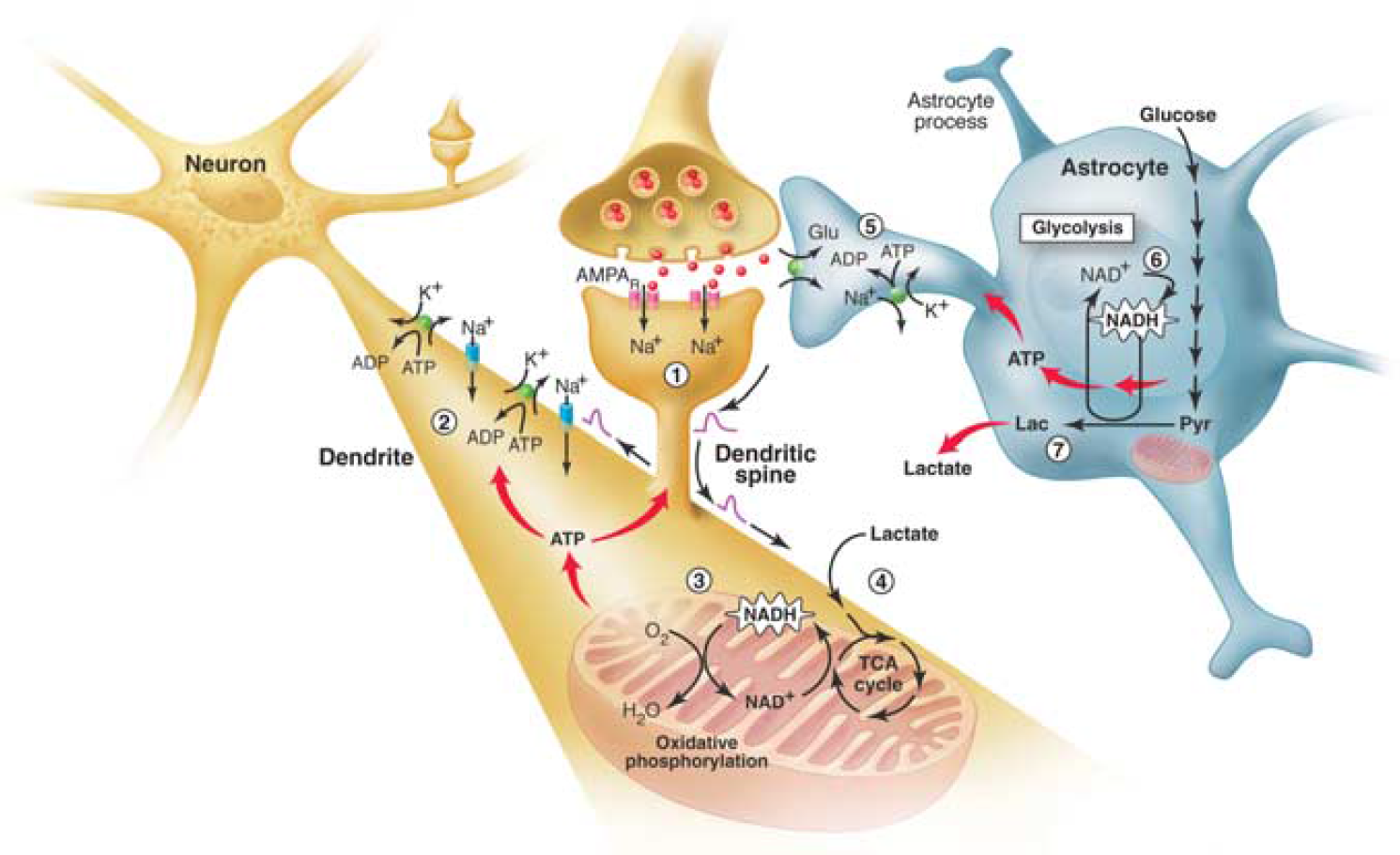
The brain energetics in the limelight. Separate activation of oxidative phosphorylation (respiration) in neurons (brown) and glycolysis in astrocytes (gray), as revealed by two-photon fluorescence imaging of nicotinamide adenine dinucleotide (NADH). (1) Stimulation of excitatory (glutamatergic) neurons activates postsynaptic AMPA receptors and induces an excitatory postsynaptic potential (EPSP) in the dendritic spine of the neuron. (2) The depolarization propagates from the dendritic spine to the dendrite, where it may cause further opening of voltage-gated sodium channels and activation of the Na+/K+ ATPase, leading to an increased demand for energy (ATP). (3) In response, oxidative phosphorylation is rapidly activated, causing a decrease in mitochondrial NADH content (the so-called ‘dip’ in the fluorescent signal). (4) Recovery of mitochondrial NADH in dendrites is accomplished by stimulation of the tricarboxylic acid (TCA) cycle, fueled largely by lactate from the extracellular pool. (5) In parallel, but delayed in time, glutamate reuptake in astrocytes (gray) activates the glial Na+/K+ ATPase. (6) The increased energy demand leads to a strong enhancement of glycolysis in the cytoplasm of astrocytes, as indicated by the large increase in cytosolic NADH fluorescence (the so-called ‘overshoot’). (7) To maintain the high glycolytic flux, NAD+ must be regenerated via the conversion of pyruvate to lactate through the activity of the enzyme lactate dehydrogenase. Release of lactate into the extracellular space not only replenishes the extracellular pool, but also may sustain the late phase of neuronal activation.
In addition to constitutive differences in the metabolic phenotype of astrocytes and neurons, metabolic responses of each cell type to external stimuli are also different. While it was never reported that neurons acutely enhanced their glucose utilization when stimulated despite several attempts to reveal it, an enhancement of glucose uptake in astrocytes mainly induced by glutamate is a widely confirmed finding now (Pellerin and Magistretti, 1994; Takahashi et al, 1995; Keller et al, 1996; Bittner et al, 2010). Concomitant to glucose utilization, modifications of glucose transport capacity induced by glutamate were observed in both astrocytes and neurons. Thus, while glucose transport was even reduced in neurons on glutamate exposure, glucose transport was conversely enhanced in astrocytes (Loaiza et al, 2003; Porras et al, 2004) (Figure 5). A critical demonstration of the preferential glucose utilization by astrocytes following synaptic activation was recently provided
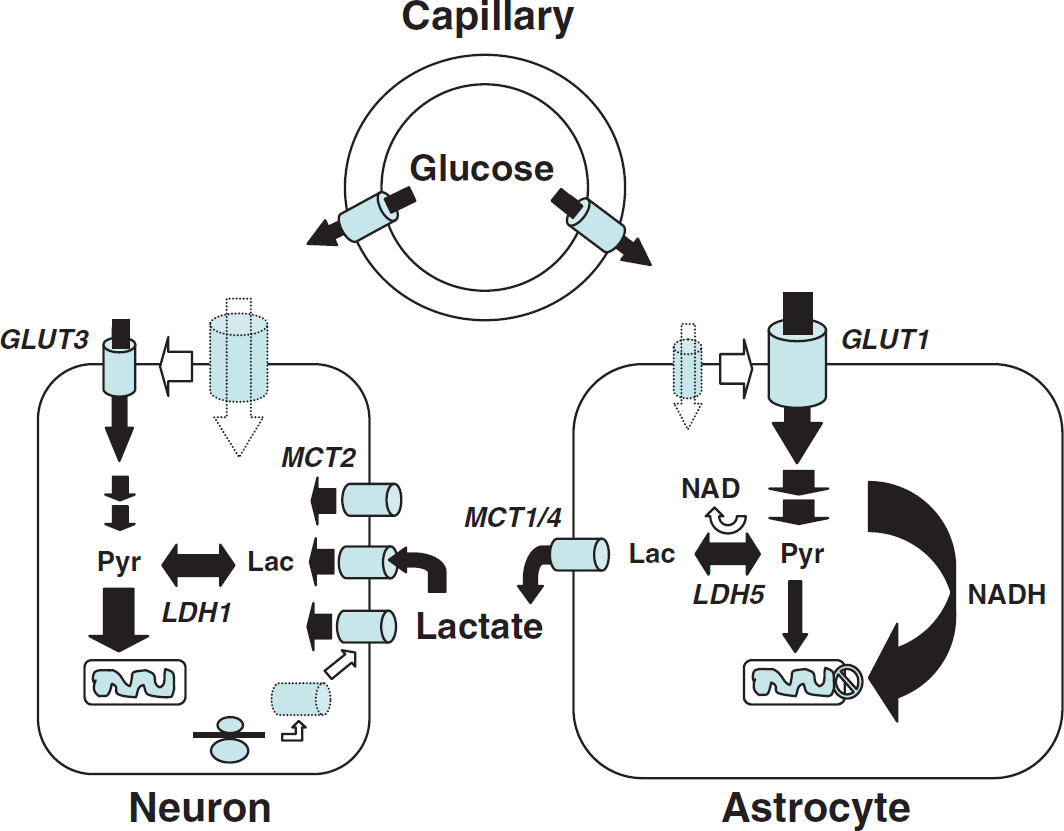
Importance of monocarboxylate transporters and the regulation of their expression/localization in lactate shuttling between astrocytes and neurons. Several unique features of astrocytes and neurons have been uncovered that suggest a partial metabolic compartmentalization and the existence of a preferential lactate transfer between the two cell types. Thus, exposure of astrocytes to glutamate was shown to directly enhance glucose transport in parallel with increased glucose utilization. Conversion of pyruvate into lactate in astrocytes is facilitated by key characteristics. Astrocytes lack a mitochondrial aspartate/glutamate carrier that reduces their capacity to transfer reducing equivalent as nicotinamide adenine dinucleotide (NADH) by the malate/aspartate shuttle in the mitochondria and regenerate NAD. To maintain the glycolytic flux, cytosolic NADH is rather converted to NAD through the reaction catalyzed by the lactate dehydrogenase isoform LDH5, preferentially expressed in astrocytes. Lactate formed is then released in the extracellular space via the high-capacity monocarboxylate transporters, MCT1 and MCT4. In contrast to astrocytes, glucose uptake in neurons is reduced by glutamate. Moreover, ascorbic acid that is shuttled from astrocytes to neurons also contributes to reduce glucose uptake in neurons. In parallel, ascorbic acid enhances lactate uptake by neurons, and lactate conversion to pyruvate is facilitated by the preferential expression of the lactate dehydrogenase isoform, LDH1. Expression of the neuronal monocarboxylate transporter, MCT2, can be enhanced through an increase in protein synthesis by various stimuli, which might contribute to long-term adaptation of energy supply to demand. GLUT, glucose transporter; Lac, lactate; LDH, lactate dehydrogenase; MCT, monocarboxylate transporter; Pyr, pyruvate. Taken from Pellerin (2008).
A Precise Molecular Description of the Astroglial-Based Coupling Mechanism Relies on Extensive In Vitro , Ex Vivo , and In Vivo Investigations
The precise mechanisms proposed to be part of the ANLS model have been the subject of several experimental investigations. First of all, the implication of glutamate transporters (and not receptors) in the glycolytic response of astrocytes has been demonstrated both
Lactate Is a Preferential Oxidative Substrate for Neurons
The consequence of glutamate-induced glucose utilization by astrocytes is the production and release of lactate in the extracellular medium immediately surrounding neurons. Curiously, a long-held view considered lactate (and more specifically lactic acid) as a toxic waste for brain cells that must be evacuated by all means from the brain parenchyma despite the fact that it is an important energy source (Dienel and Hertz, 2001). However, evidences accumulated over >50 years have clearly established that lactate represents one of the rare alternative oxidative substrate (together with ketone bodies) for neurons (McIlwain, 1953; Pellerin, 2003). Moreover, in the presence of both substrates as it is the case physiologically, it was shown that lactate is largely preferred as an oxidative energy substrate over glucose by neurons (Itoh et al, 2003; Bouzier-Sore et al, 2003, 2006; Ivanov et al, 2011), although glucose utilization also occurs in neurons (reviewed in Mangia et al, 2009). Prominent lactate utilization by the brain and more specifically by neurons has also been confirmed
Opening the Shuttle's Doors: Key Role of Monocarboxylate Transporters
The possibility of shuttling lactate between brain cell types is determined by the expression of specific transporters exhibiting different kinetics. Indeed, modeling studies have demonstrated the key role that monocarboxylate transporters play in regulating lactate influx and efflux (Aubert et al, 2005). In recent years, a precise description of the expression and distribution of monocarboxylate transporters in the central nervous system has been provided by immunohistochemical studies (reviewed in Pierre and Pellerin, 2005). Thus, the three main proton-dependent lactate carriers known as MCT1, MCT2, and MCT4, have been identified and extensively studied both
The distribution of monocarboxylate transporters is cell specific. MCT1 is expressed on endothelial cells forming blood vessels as well as on astrocytes and oligodendrocytes (Gerhart et al, 1997; Rinholm et al, 2011). MCT2 appears predominantly present in neurons, where it is found on axons and dendrites, with a particular enrichment in dendritic spines (Bergersen et al, 2001; Pierre et al, 2002). In contrast, MCT4 is exclusively expressed by astrocytes with a polarized distribution showing a more important concentration on processes in contact with synapses (Rafiki et al, 2003; Pellerin et al, 2005). It is interesting to consider the kinetic properties of these transporters. MCT2 is a high-affinity transporter with a Km for lactate of 0.7 mmol/L. MCT1 and MCT4 have a much lower affinity for lactate (Km for lactate of 3.5 and 34 mmol/L, respectively). Their cell-specific distribution is paralleled by an enriched expression of particular LDH isoforms displaying different kinetic characteristics. Isoforms containing the LDHB subunits are more abundant in neurons while isoforms enriched in LDHA subunits are enriched in astrocytes (Bittar et al, 1996; O’Brien et al, 2007). While the main determinant of the lactate:pyruvate equilibrium is the redox state and the suggestion has been made that ‘despite changed kinetic constants, the equilibrium constant, Keq, is the same for all isoenzymes, because it is the same chemical reaction being catalyzed’ (quoted from: Quistorff and Grunnet, 2011), the association of the high-affinity transporter MCT2 with LDH isoforms enriched in LDHB subunits creates kinetic conditions highly favorable for lactate uptake and utilization by neurons. The presence of LDH isoforms predominantly exhibiting LDHA subunits together with the high-capacity monocarboxylate transporters MCT1 and MCT4 rather provide optimal settings to favor lactate production and release from astrocytes. Considering the preferential metabolic profile exhibited by each cell type as described above, these characteristics reinforce the concept that lactate is shuttled preferentially from astrocytes to neurons, with varying degree depending on the activation state (Figures 6A and 6B).
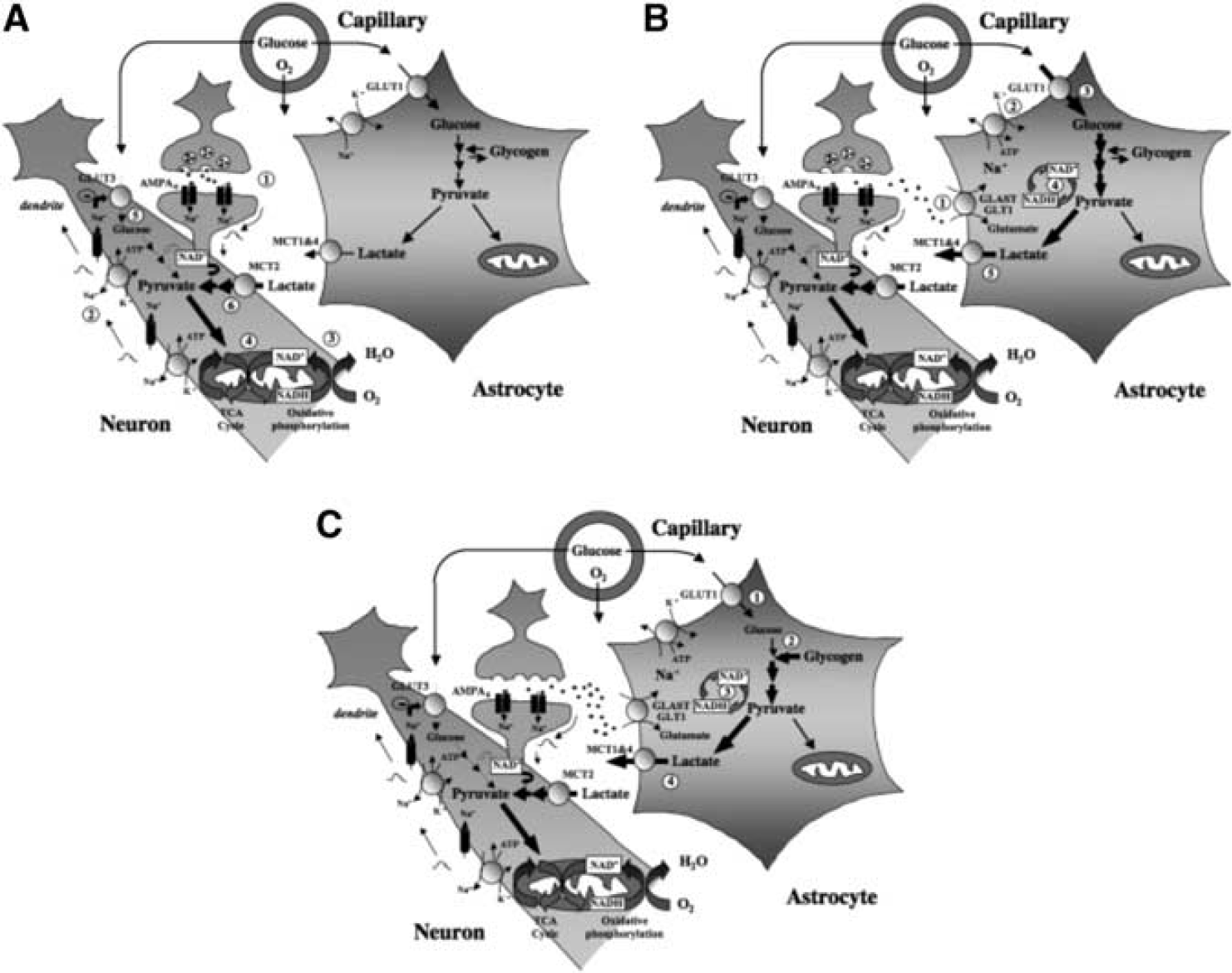
Contribution of glucose- and glycogen-derived lactate to support different phases of neuronal activation. (
Energy substrate utilization can be limited by the transport capacity. It was previously shown that this could be the case for neuronal lactate utilization since MCT2 overexpression in cultured neurons using viral vectors promoted lactate utilization in these cells when they were stimulated with kainate (Bliss et al, 2004). It became of interest to determine whether the expression of monocarboxylate transporters could be regulated in the brain cells. Indeed, it was found that MCT2 expression in neurons was under regulation. Noradrenaline (NA), insulin, insulin-like growth factor-1 (IGF-1), and brain-derived neurotrophic factor (BDNF) were all shown to enhance MCT2 expression in cultured neurons (Chenal and Pellerin, 2007; Chenal et al, 2008; Robinet and Pellerin, 2010). The mechanism involves a stimulation of translation via the phosphatidylinositol 3-kinase/serine-threonine protein kinase from Akt virus/mammalian target of rapamycin (PI3K/Akt/mTOR/S6) pathway. Moreover, it was shown that such an activation occurs at the synaptic level (Robinet and Pellerin, 2010). In addition to changes in the overall expression levels, evidence was provided that the amount of MCT2 proteins present at the cell surface can be modified (Figure 5). It was shown that translocation of MCT2 from an endogenous pool to the cell surface can be induced in cultured cortical neurons by exposing them to a combination of glutamate and glycine (Pierre et al, 2009). Quite importantly, it was shown that a doubling of MCT2 expression at the cell surface leads to an increase of ∼80% in lactate transport (Pierre et al, 2009). Thus, changes in expression and localization of MCT2 induced by neuroactive signals cause significant modifications of neuronal energetics. Since such adaptations appear to occur quite rapidly (<1 minute measured with static approaches), it is likely that they represent the most appropriate process by which neurons could adapt their substrate supply and concomitant energy production to face changing needs coming with fluctuating activity. It is expected that in the case of synaptic plasticity, changes in synaptic efficacy will be accompanied by adaptations in energy supply. The AMPA type glutamate receptor subunit GluR2 is known to participate to the mechanism of synaptic plasticity at glutamatergic synapses (Isaac et al, 2007). Observations that MCT2 not only interacts with GluR2 but modifies its subcellular distribution and expression levels support the idea that energy supply might be tightly regulated as part of the mechanism of synaptic plasticity (Maekawa et al, 2009; Pierre et al, 2009).
Glycogen: the Astrocytic Energy Reserve Essential for Neurons
One of the defining features of the ANLS is the postulated central role of lactate transfer from astrocytes to neurons. In addition to glutamate-stimulated aerobic glycolysis, another astrocyte-based metabolic pathway can produce lactate, namely glycogenolysis. Glycogen is almost exclusively localized in astrocytes (Magistretti, 2008). Glycogen is not found in adult neurons, except occasionally in large neurons in the brainstem or in the peripheral nervous system (Sotelo and Palay, 1968), despite the fact that quite surprisingly, glycogen synthase—the enzyme responsible for glycogen synthesis—is present in many neurons. However, two active mechanisms operate to inhibit glycogen storage in neurons. First, a proteasome-dependent mechanism, involving the malin–laforin complex, is active in neurons to permanently degrade glycogen synthase (Vilchez et al, 2007). Second, activity of the residual glycogen synthase is inhibited by phosphorylation. The reasons for such a complex regulation are unknown, but certainly vital for neurons: indeed, manipulations aimed at allowing glycogen synthase activity and hence glycogen synthesis in neurons, lead to neuronal glycogen accumulation and apoptosis. For example, a loss-of-function mutation in the malin–laforin complex, characterized by accumulation of glycogen-containing deposits in neurons, results in progressive myoclonus epilepsy, also known as Lafora disease.
As mentioned earlier, in neurons, glycolysis is reduced to a minimum, due to the constitutive proteasome-dependent degradation of the glycolysis-promoting enzyme 6-phosphofructo-2-kinase/fructose 2,6-bisphosphatase, isoform 3, PFKFB3 (Herrero-Mendez et al, 2009). In these cells, glucose metabolism is predominantly directed to the oxidative branch of the pentose phosphate pathway, resulting in production of reducing equivalents necessary to maintain the protective antioxidant status of neurons. Removal of PFKFB3 inhibition, resulting in increased glycolysis leads to oxidative stress and apoptotic death of neurons. Taken together, these data indicate that neurons can simply not afford to process glucose through the glycogen cycle (Magistretti and Allaman, 2007); furthermore, upregulation of glucose processing through glycolysis in neurons can lead to death, thus providing a metabolic contraindication for these cells to produce lactate. As reviewed earlier, this is in stark contrast to the metabolic phenotype of astrocytes in which glycogenolysis and glycolysis, both leading to lactate production (Dringen, 2000; Pellerin and Magistretti, 1994), are the preferred metabolic pathways for glucose. These considerations imply that the ANLS can also be operated by promoting glycogenolysis in astrocytes in response to signals released by active neurons (Figure 6C).
A decade before the glutamate-stimulated aerobic glycolysis was described, it had been shown that a restricted number of neurotransmitters/modulators could promote glycogenolysis (Magistretti et al, 1981; Magistretti and Morrison, 1988). The glycogenolytic agents included NA, vasoactive intestinal peptide (VIP), and adenosine. This metabolic action of VIP, which in the neocortex is contained in locally acting bipolar neurons and hence restricted to cortical columnar modules, was contrasted to that of NA, which given the horizontal orientation of noradrenergic intracortical fibers that span horizontally across functionally distinct cortical areas (Magistretti and Morrison, 1988), would be in a position to prime metabolically the neocortex globally (Magistretti and Morrison, 1988) (Figure 7). Later on, the existence of ‘metabolic hotspots’ was described in the neocortex as a result of the synergistic interaction between VIP and NA at the levels of cortical columns (Magistretti and Schorderet, 1984).
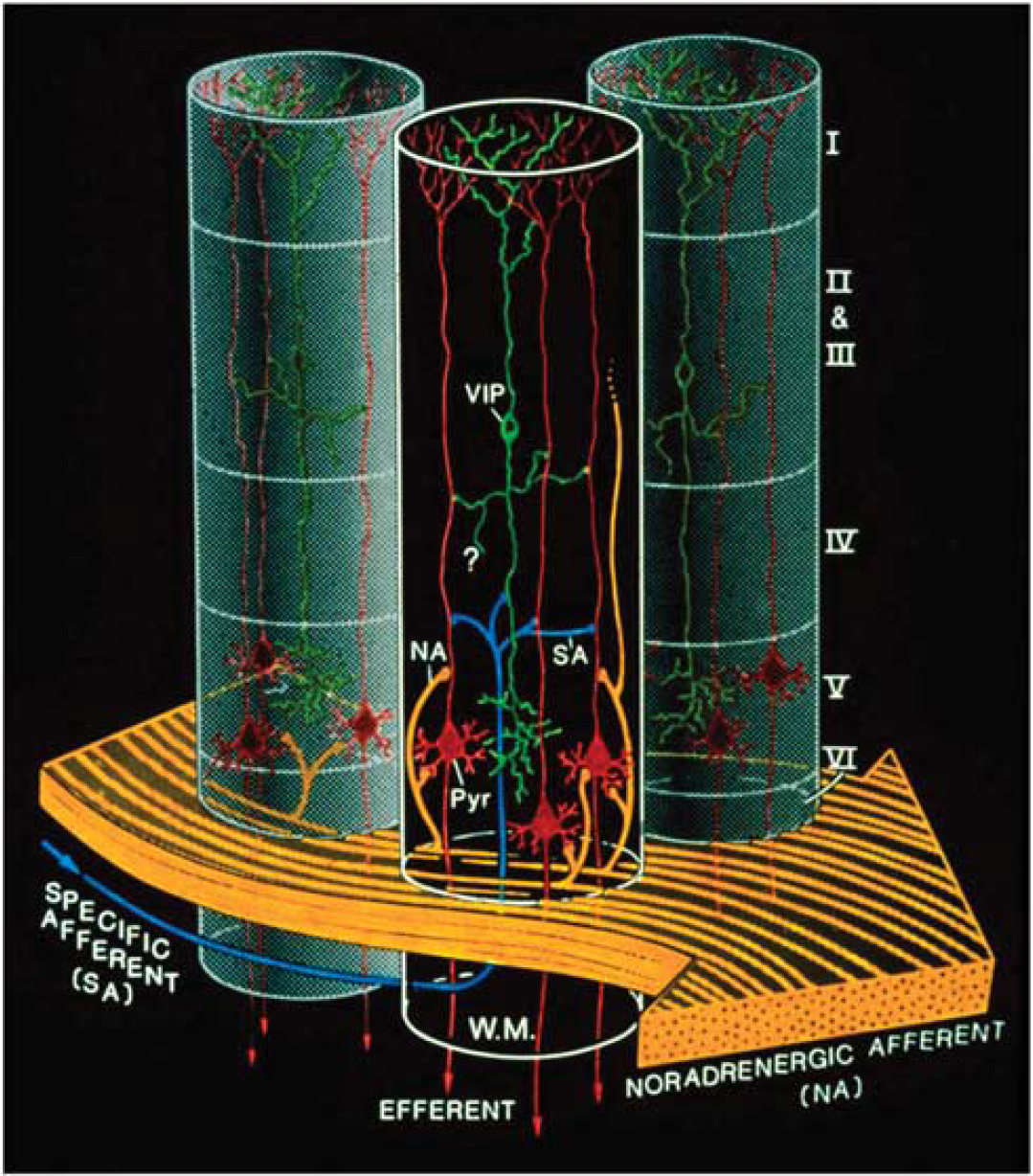
Complementary spatial domains in which vasoactive intestinal peptide (VIP)-containing bipolar neurons and noradrenergic fibers exert their glycolytic effects. Such an arrangement would allow for the establishment of temporary columnar ‘hotspots.’ VIP, VIP-containing bipolar cell; NA, noradrenergic afferents; Pyr, pyramidal cell furnishing major efferent projections; SA, specific afferent (i.e., from thalamus or from other cortical areas); WM, subcortical white matter. Cortical layers denoted by roman numerals. Note tangential orientation of NA fibers, and radially restricted domain of VIP-containing neuron. The concomitant activation of noradrenergic fibers (large, yellow arrow, bottom) by unexpected sensory stimuli and of a group of VIP-containing intracortical neurons by specific thalamic inputs (small, blue arrow, bottom left) would lead to a drastic increase in cAMP levels within a discrete volume of somatosensory cortex, delineated here by the cylinder with dark background. Adjoining gray-background cylinders represent nonactivated cortical volumes. For graphic clarity, only the VIP-containing (green) and the pyramidal cells (red) have been represented here. In particular, the principal target cells of the thalamocortical afferents, that is, the small stellate cells in layer IV, have been omitted. However, any cell with the capacity for dendritic reception in layer IV may receive thalamic inputs. Furthermore, also for graphic clarity, only one VIP neuron per cylinder has been drawn, in representation of a group of VIP neurons. This is a heuristic model, and as such, certain details of the synaptic circuitry depicted in the diagram (e.g., direct thalamocortical input to VIP-containing cells) are suggested by presently available data, but have not been demonstrated definitively. However, all aspects of the model are potentially testable. Taken from Magistretti and Morrison (1988).
In summary, lactate can be produced by astrocytes following two kinds of neuronal signals: glutamate-stimulated aerobic glycolysis and glycogenolysis triggered by VIP, NA, or adenosine (Magistretti and Morrison, 1988; Magistretti et al, 1986). As far as glycogenolysis is concerned, its function in neurometabolic coupling has been considered to be a sort of general mechanism that would provide additional energy substrates to match increases in neuronal activity (Magistretti, 2008). Thus, stimulation of the whisker-to-barrel pathway was shown to result in glycogenolysis in the barrel field of layer IV somatosensory cortex (Swanson et al, 1992). Maintenance of sustained axonal spiking in mouse optic nerve was shown to depend on adequate glycogen content and lactate shuttling between astrocytes and the axonal fibers (Tekkök et al, 2005). Very recently, a specific role of astrocytic glycogenolysis and the ensuing astrocyte–neuron lactate transfer has been demonstrated for synaptic plasticity in a learning paradigm (Suzuki et al, 2011).
Thus, pharmacological inhibition of glycogenolysis in the hippocampus with the glycogen phosphorylase inhibitor DAB (Walls et al, 2008), results in loss of long-term memory in an inhibitory avoidance learning paradigm. A similar result is obtained by disrupting the expression of the astrocyte-specific lactate transporter MCT4. These memory-inhibiting effects are fully rescued by local administration of lactate but not by equicaloric glucose. Disruption of the expression of the neuron-specific lactate transporter MCT2 also leads to amnesia, which however is not rescued by exogenous lactate, indicating that lactate import into neurons is necessary for long-term memory formation. In keeping with the behavioral effects observed, the
Some Controversies
While there is a considerable support, both theoretical and experimental, for the existence of an activity-dependent transfer of lactate between astrocytes and neurons (see preceding paragraphs), there are still a few diverging views that dispute some aspects of the ANLS. Thus, one argument derived from modeling studies suggests that the glucose transport capacity is larger in neurons than in astrocytes (DiNuzzo et al, 2010a; Mangia et al, 2009; Simpson et al, 2007), a prediction suggested to be inconsistent with ANLS. This theoretical conclusion is at odds with the observation that haploinsufficient mice for the neuron-specific glucose transporter GLUT3 do not present any neurologic or brain energy metabolism phenotype, and in particular do not display any decrease in brain glucose utilization as determined by fluorodeoxyglucose micro-PET (Schmidt et al, 2008; Stuart et al, 2011). In contrast, haploinsufficient mice for the glucose transporter GLUT1, which is absent in neurons, present a severe neurologic phenotype of impaired motor activity and coordination, microencephaly and frequent seizures (Wang et al, 2006).
Furthermore, in a recent article (Jolivet et al, 2010), we have pinpointed some major flaws in the modeling work proposed by DiNuzzo et al (2010a) and Simpson et al (2007). Thus for example, the biological data used for the modeling concerning the density of glucose transporters in astrocytes and neurons were obtained by measuring cytochalasin B binding in cultures. Using such data obtained from semiquantitative analysis of transporter density in cultures to model quantitative
Using an elegant cell-specific neurochemical technique, Gandhi et al (2009) have shown that when raising artificially extracellular lactate concentrations up to 40 mmol/L in brain slices, astrocytes display a higher transport and distribution capacity for lactate compared with neurons. From this observation, they conclude that a neuron-to-astrocyte shuttling is more likely than the opposite. The results of these experiments are not surprising: indeed astrocytes, as shown by the authors and many other investigators, are tightly coupled through gap junctions, a cytological organization that produces a large ‘sink’ for lactate disposal when compared with single, isolated neurons. Thus, increasing artificially extracellular lactate, quite expectedly will result in its uptake into both cell types, but with a significant higher capacity in astrocytes because of the considerable greater size of the astrocytic network compared with isolated neurons. Furthermore, and possibly more importantly, experiments were carried out in the absence of stimulated neuronal activity, thus resulting in conditions in which the energetic needs of neurons are not engaged. Under conditions in which the ANLS would be operational, the source of extracellular lactate is the astrocytes, a fact that would determine the directionality of the lactate flux, as shown for example by the study of Rouach et al (2008).
The fact that lactate could be a valuable energy substrate capable to sustain neuronal activity in brain slices was recognized already 60 years ago by McIlwain (1953), and later confirmed by various authors including in particular Schurr et al (1988) and Izumi et al (1997). This role of lactate as a substrate in slices maintained
Another modeling study by DiNuzzo et al (2010b), commented by Dienel (2010) has suggested that glycogenolysis in astrocytes provides a mechanism to preserve glucose availability for neurons, rather than providing lactate to them. Experimental data obtained
Finally, a critique that has been raised, particularly at the early times of the formulation of the ANLS model, was that it was largely based on
Conclusion
The ANLS model has now reached maturity. It is ready to settle in different domains of neurosciences well beyond the restricted circle of brain energy metabolism aficionados. Moreover, it bears a heuristic value to approach fundamental functions such as neuroprotection and memory (Figure 8). In doing so, it repositions neuroenergetics as a central player in brain function and information processing. After a healthy (and hectic at times!) development, the ANLS is ready to engage in exciting new ventures.
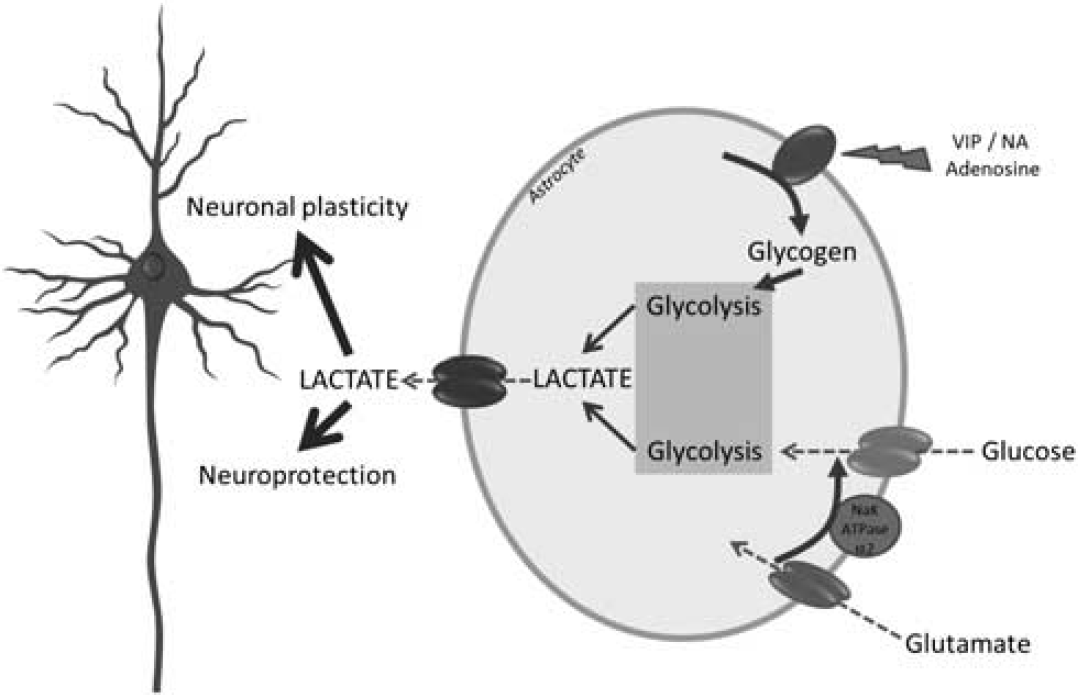
Roles of glucose- and glycogen-derived lactate in neuroprotection and neuronal plasticity. It is suggested that under different levels of brain activation, glutamatergic as well as specific neuromodulatory systems (e.g., noradrenergic) will be activated. Glutamate reuptake in astrocytes will trigger glucose-derived lactate production and release. In parallel, neuromodulators such as vasoactive intestinal peptide (VIP), noradrenaline (NA), or adenosine will stimulate glycogenolysis, leading as well to lactate production and release. Such lactate provision to neurons by astrocytes turns out to be essential for the establishment of memory via support of synaptic plasticity processes as well as for neuroprotection of neurons under certain stress conditions (e.g., excitotoxicity).