
Abstract
While communicating hydrocephalus (CH) is often characterized by increased pulsatile flow of cerebrospinal fluid (CSF) in the cerebral aqueduct, a clear-cut explanation for this phenomenon is lacking. Increased pulsatility of the entire cerebral vasculature including the cortical capillaries has been suggested as a causative mechanism. To test this theory, we used two-photon microscopy to measure flow pulsatility in neocortical capillaries 40 to 500 μm below the pial surface in adult rats with CH at 5 to 7 days (acute,
Introduction
While it is well known that cerebrospinal fluid (CSF) flow in the cranium has a marked cardiac-induced pulsatile component, whether CSF pulsatility has an important role in disease remains a topic of ongoing debate. A number of studies have linked alterations in pulsatile CSF flow with various pathologies, such as hydrocephalus (Bradley et al, 1996; Luetmer et al, 2002; Wagshul et al, 2009), syringomyelia (Mauer et al, 2008), and Chiari malformations (Alperin et al, 2005; Quigley et al, 2004), but these associations do not give any indication for a causative role of CSF pulsatility in these conditions. On a more fundamental level, while elevated CSF flow pulsations in the cerebral aqueduct have been shown in numerous studies, mostly in normal pressure hydrocephalus (Bateman and Loiselle, 2007; Bradley et al, 1996; Kahlon et al, 2007; Luetmer et al, 2002; Miyati et al, 2003), the exact mechanism for elevated pulsatility is still debated. Until the driving mechanism behind elevated CSF flow pulsatility is well understood, it is uncertain how this potential marker of disease and recovery in hydrocephalus can serve as an acceptable prognostic tool.
It has been suggested that the increase in CSF pulsatility in communicating hydrocephalus (CH) stems from changes in the compliance of the subarachnoid space, leading to a redistribution of the subarachnoid space CSF flow pulsatility into the brain microvasculature (Greitz, 1993). This would explain the increase in macroscopic pulsations in the cerebral aqueduct, because of the mechanical coupling between the ventricular walls and the parenchyma. However, a potentially more significant implication of this theory would be an overall increase in vascular pulsations throughout the brain, including the cerebral microvasculature. Increased microvascular pulsatility can have pathological implications, because increased pulsatile stress forces can activate potent vasoactive factors (Bilfinger and Stefano, 2000; Silacci et al, 2001; Ziegler et al, 1998). However, the primary counter theory suggests that increased CSF pulsations are simply a product of the reduced intracranial compliance, due to either increased intracranial pressure or tissue compression from the expanded ventricles, resulting in elevated brain pulsations as dictated by the exponential pressure—volume curve (Marmarou et al, 1975). This theory would imply that while pressure pulsations in the brain may be increased in hydrocephalus, these may not be accompanied by any change in microvascular flow pulsatility.
Thus, the primary purpose of this study was to test the theory that pulsations in microvascular flow are elevated in hydrocephalus, and that these are associated with elevated aqueductal flow pulsations. We used two-photon laser-scanning microscopy (Denk et al, 1990; Kleinfeld et al, 1998) to quantify microvascular flow pulsations
Materials and methods
All experimental procedures were performed in accordance with the NIH Guide for Care and Use of Laboratory Animals, and approved by the Institutional Animal Care and Use Committee of Stony Brook University (Stony Brook, NY). Every effort was taken to minimize the discomfort and the number of animals used.
Communicating Hydrocephalus Induction and Study Design
Twenty-five female Sprague-Dawley rats (200 to 250 g) were included in this study. Communicating hydrocephalus was induced in 16 rats by injecting kaolin into the basal cisterns, using the technique developed by us (Li et al, 2008); nine animals served as controls. Postoperatively, animals exhibiting lethargy and weight loss received subcutaneous supplements of 5% dextrose and Ringer's lactate for 3 to 5 days. Multiphoton microscopy was performed at either the acute (5 to 7 days after induction,
Two-Photon Microscopy
An optically transparent cranial window was opened in the right parietal bone to provide access to neocortical capillaries for imaging. Each animal was anesthetized with an intraperitoneal injection of ketamine/xylazine (70/7 mg/kg) and secured to a custom-made head holder with ear bars. The head was shaved, the skin incised and a 5 × 3 mm craniectomy, centered ∼5 mm posterior and 2.5 mm lateral to Bregma, was made by thinning the bone along the periphery and then carefully removing the bone flap. The drilling area was irrigated frequently with cold saline to prevent heating of the underlying tissue; the dura was left intact in all animals. A styptic pad was gently applied to the dura to stop any bleeding. The craniectomy was then filled with warm 1% agarose and covered with a custom-cut glass coverslip, which was then sealed to the skull with cyanoacrylate gel. The sealed cranial window served to maintain normal intracranial pressure and flow dynamics. A metal plate with a 3 × 1 cm opening surrounding the craniectomy was attached to the skull with cyanoacrylate gel and screwed to the head holder to prevent further head motion.
Multiphoton imaging was performed on an Olympus BX60WI microscope (Olympus America Inc., Center Valley, PA, USA), equipped with a 1.7-W Chameleon laser (Coherent Inc., Santa Clara, CA, USA), and a 60 × water immersion objective. After the animal was positioned under the microscope, 0.5 mL of 70 kDa fluorescein isothiocyanate dextran (Sigma, St Louis, MO, USA), dissolved in 1 mol/L PBS at 40 mg/mL, was injected through the tail vein. The fluorophore labels blood serum but is not taken up by red blood cells (RBCs), causing them to appear as dark moving objects against the bright, labeled serum. Optimal two-photon excitation was at 800 nm.
Capillaries were imaged at 2 to 4 nonoverlapping sites within the cranial window with a total of 20 to 40 capillaries in each animal, at depths of 40 to 500 μm; all depths were measured from the pial surface. They exclude the thickness of the dura and cover cortical layers 1 to 3; the dorsal edge of layer 4 is ∼500 μm from the pial surface in control rats. Capillaries were visualized by 2D planar scans (Figure 1A), which were later used for vessel diameter determination. Capillaries selected for flow measurements had a straight in-plane length of at least 10 μm. Individual line scans were ∼16 seconds long with temporal resolution of 1.6±0.3 ms per line and spatial extent of 26±4 μm (Figure 1D). The microscope was controlled by FluoView FV300 software (Olympus Inc.) running on a desktop PC.
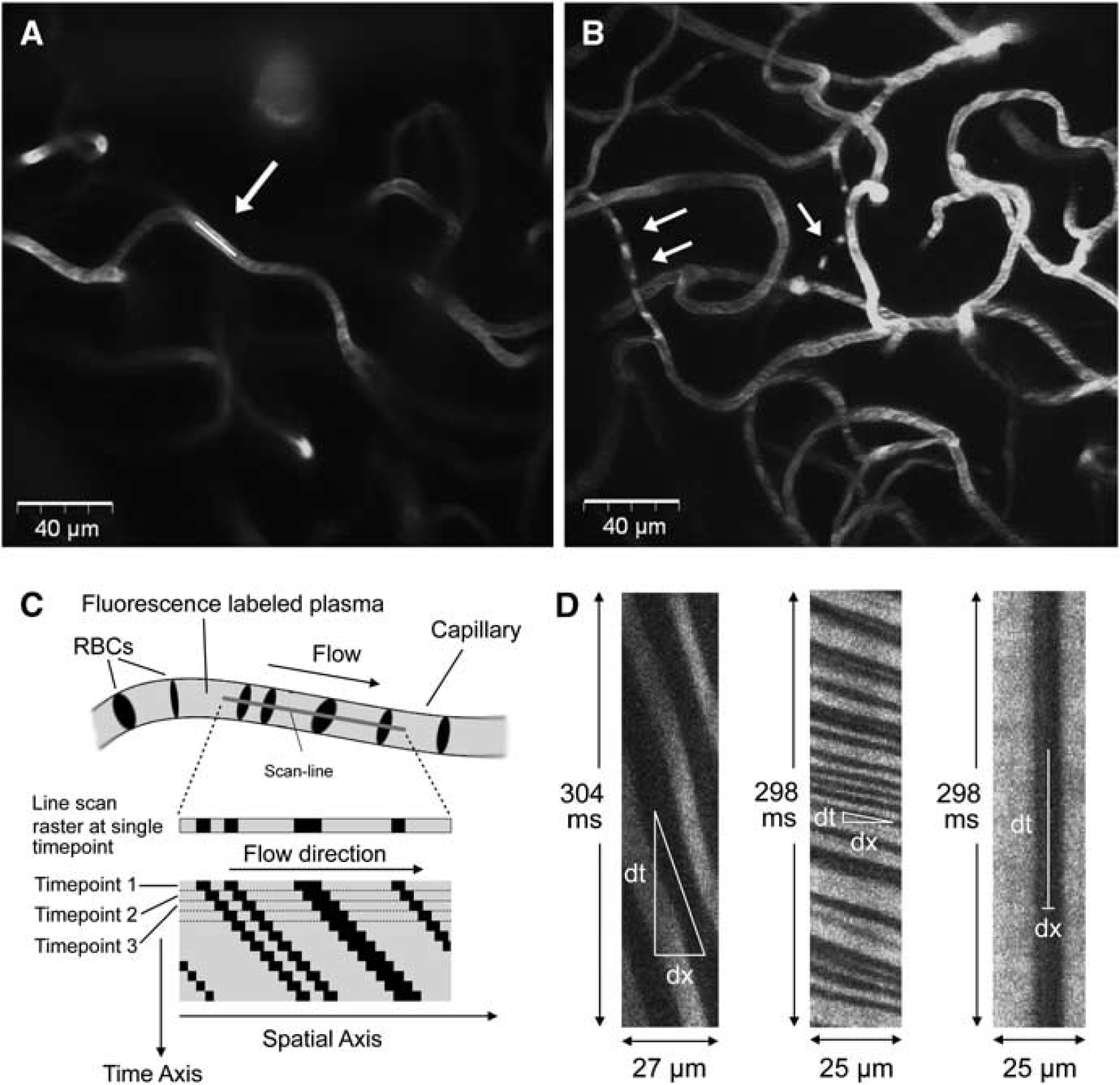
A 2D planar image showing a section of a capillary network, at a depth of 160 μm, in a control animal. The white line indicates the line over which the capillary was scanned (
Anesthesia was maintained throughout imaging with 50% supplements of the initial ketamine/xylazine dose, as needed. Electrocardiogram pads were attached to the animal's fore and hind paws, and the electrocardiogram was recorded continuously for anesthesia monitoring and heart rate (HR) determination in the data processing. Animals were euthanized at the end of the imaging session by an overdose of pentobarbital.
Image Processing
All image processing and data analysis were performed using Matlab applications (The Mathworks, Natick, MA, USA). Moving RBCs form dark bands in the line-scan image (Figure 1C) so that RBC velocity at any given point on the image is related to the slope of the bands. To calculate the velocity as a function of time, the line-scan image was broken into small time segments of ∼45 ms, which were overlapped to produce an effective temporal resolution of ∼15 ms per data point. Slopes were calculated for each segment using the singular value decomposition method first described by Kleinfeld et al (1998), as follows: the segment was rotated incrementally through a range of angles, and a parameter
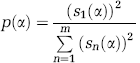
where
Line-scan images of poor quality (e.g., RBC bands not visually distinguishable) and images with severe motion artifacts were excluded from analysis. Single-point spikes in the velocity waveform, usually due to either small motion artifacts or low RBC density, were corrected automatically with a median filter; data sets with <4 contiguous seconds of spike-free velocity data were discarded.
Pulsatility Index
Pulsatility was quantified from the velocity waveforms with the Gosling PI, the ratio of the mean peak-to-peak amplitude of the waveform to its mean velocity, typically calculated in the time domain (Gosling and King, 1974). However, to limit pulsatility calculations to cardiac-induced pulsations, PI was computed in the frequency domain from the power spectrum of the velocity waveform by integrating over the cardiac frequency peak and its second harmonic, which were identified using the animal's HR extracted from the electrocardiogram waveform. This integral was then divided by the first bin of the power spectrum, which equals the mean velocity, to produce the PI. A baseline, computed from the average of the spectral points immediately adjacent to the peaks, was subtracted before PI calculation to remove the effects of background noise.
Calculation of Vessel Diameter
Capillary diameters were measured from 2D planar scans by selecting a profile perpendicular to the axis of the capillary, averaged over the segment through which flow was measured. A flat-topped function with Gaussian edges was fitted to this profile, and diameter was measured as the full-width at half-maximum of the function.
Statistical Analyses
Differences in vessel diameter, vessel depth, and HR between the three experimental groups (acute CH, chronic CH, and controls) were compared using one-way repeated measures analysis of variance.
Pulsatility indices were compared among the three experimental groups using two linear mixed models, treating repeated measures in each animal as a random effect and controlling for vessel diameter, vessel depth, and HR, all of which varied randomly within the data sets. In one model, fixed effects of experimental group, diameter, depth, and HR on PI were tested. In a second model, three interaction terms group by depth, group by diameter, and group by HR were added to the model to test for differences within the groups based on linear variations in PI with these varying experimental parameters.
Based on the results of these initial models showing significant group by depth and group by HR interactions,
Pearson correlations were used to examine the association between capillary PI (calculated for each animal, averaged over all vessels) and aqueductal SV. Similarly, Pearson correlations between PI and VV were also used to determine the relationship between capillary pulsatility and ventricular dilation.
All data are reported as mean±standard deviation. Normal distribution of data was verified using histograms. Statistical computations were performed in SAS (SAS Institute Inc., Cary, NC, USA), and results were considered significant at
Results
Communicating Hydrocephalus Induction
Three chronic animals did not develop hydrocephalus and were excluded from analysis. Figure 2 illustrates the marked difference in ventricular size between hydrocephalic and control animals. Mean VV was 132±31 and 77±31 μL for the acute and chronic groups, respectively; the difference between these groups was significant (
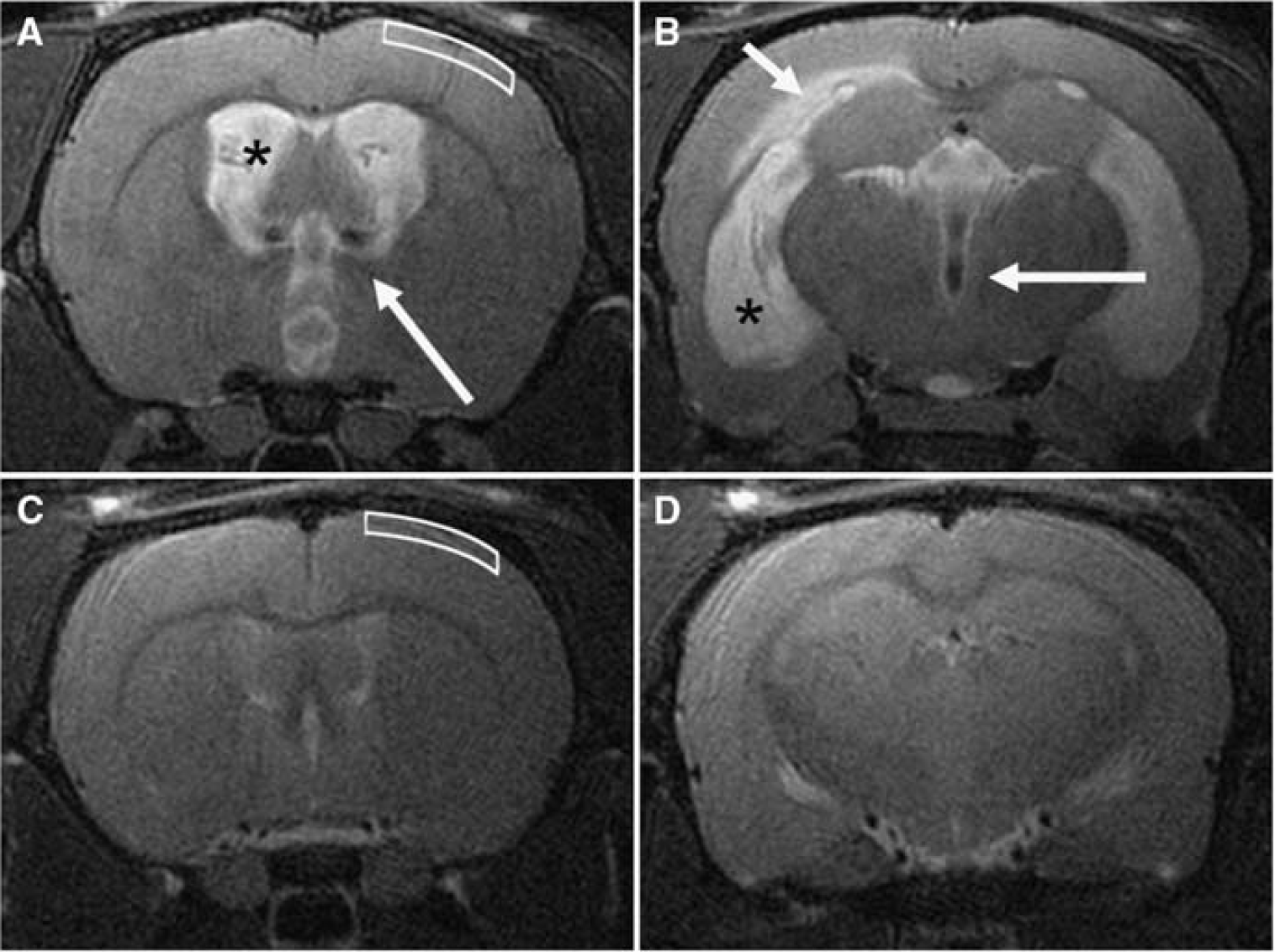
Representative T2-weighted coronal magnetic resonance images at the level of the foramen of Monro (
Two-Photon Microscopy
Nineteen percent of line-scan images were discarded due to the presence of artifacts, poor quality, or abnormal flow. In some images, cardiac- or respiratory-induced pulsations caused excessive vessel motion and periodic motion artifacts in the line-scan images. Images with very low RBC density resulted in large data segments with no RBCs, and thus no recognizable features for the velocity calculation. This was the primary cause of spikes in the extracted velocity waveform, and as noted above, data sets with <4 seconds of continuous RBC motion were excluded. In some vessels, blood flow in a capillary slowed or stopped, and then resumed after a few seconds or even minutes. In rare cases, flow would reverse direction. In images with such artifacts or aberrant flow, the portion of the image containing the artifact(s) or arrested flow was discarded, and only the portion of the image with good contrast was used for velocity waveform analysis.
Valid RBC velocity waveforms were collected in 765 capillaries (323 acute CH, 184 chronic CH, and 258 intact controls). Average depth of capillaries for all animals was 254±90 μm beneath the pial surface; all capillaries were deeper than 40 μm, with one third deeper than 300 μm, and the deepest capillary was imaged at 549 μm. The maximum depth achieved in each animal depended mainly on the quality of the cranial window. Because the dura was left intact, capillaries could not be visualized in most animals beneath 450 μm. Mean capillary depths for the acute, chronic, and control groups were 276±78, 259±98, and 242±99 μm, respectively; the depth for the control group was significantly smaller than that of the acute group (
Capillary Pulsatility
Most waveforms were clearly pulsatile, with a distinct frequency peak at the electrocardiogram-derived HR. Two representative examples of line-scan images, and their associated velocity waveforms and frequency spectra, are shown in Figure 3.
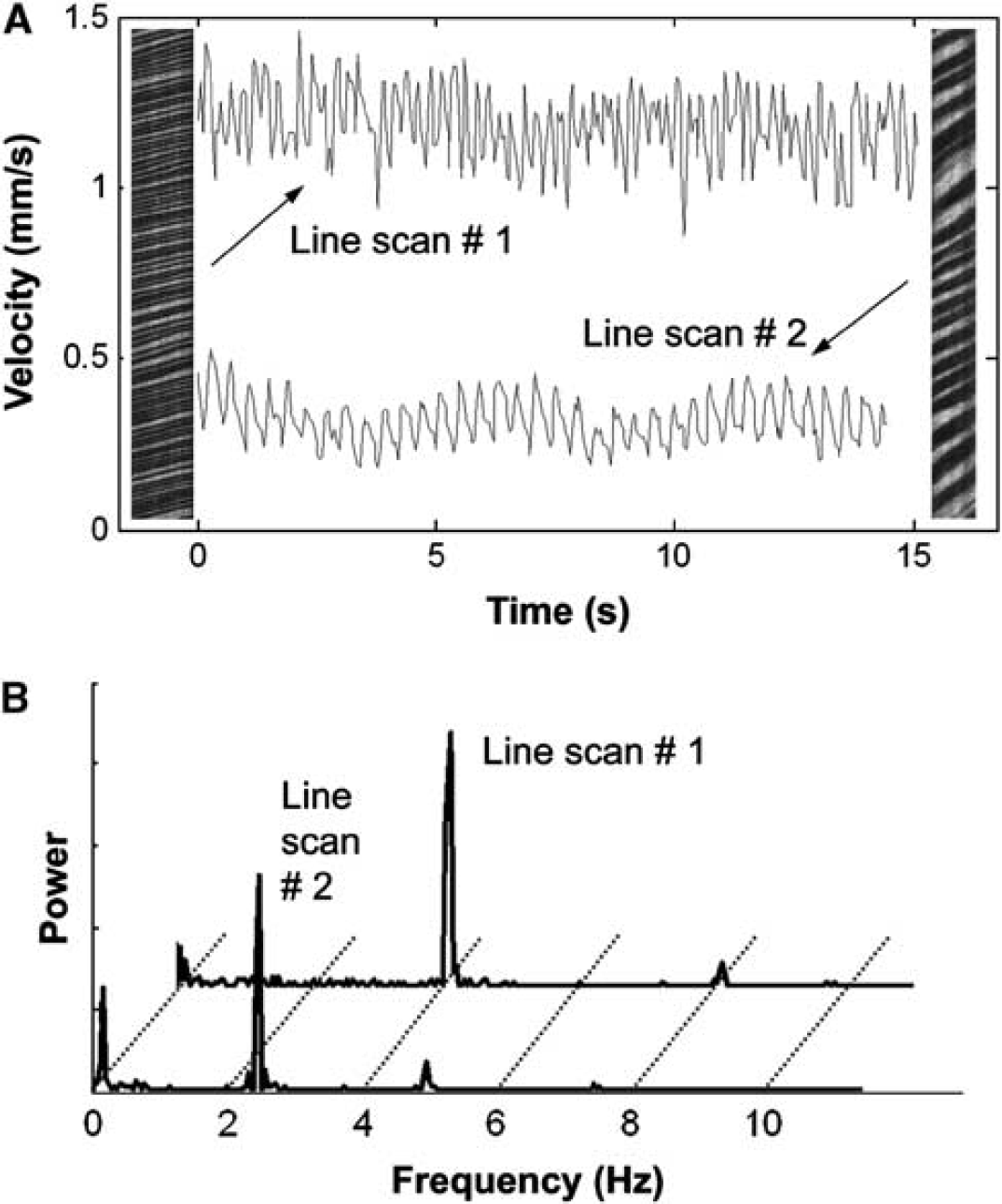
Two representative line scans with their corresponding velocity waveforms (
The highly significant, linear relationship between absolute pulse amplitude and mean flow velocity for all measurements in the study, illustrated in Figure 4, provides good justification of the choice of PI, the ratio of pulse amplitude to mean velocity, as a measure of flow pulsatility which is minimally influenced by variations in vessel caliber and mean flow velocity. Pulsatility indices for the acute, chronic, and control groups, as analyzed in the first linear mixed model without controlling for HR, depth or vessel size, are shown in Figure 5. Pulsatility indices were 0.15±0.06 (acute), 0.14±0.05 (chronic), and 0.18±0.07 (control); although there was a trend toward lower PIs for the hydrocephalic animals, this was not statistically significant (
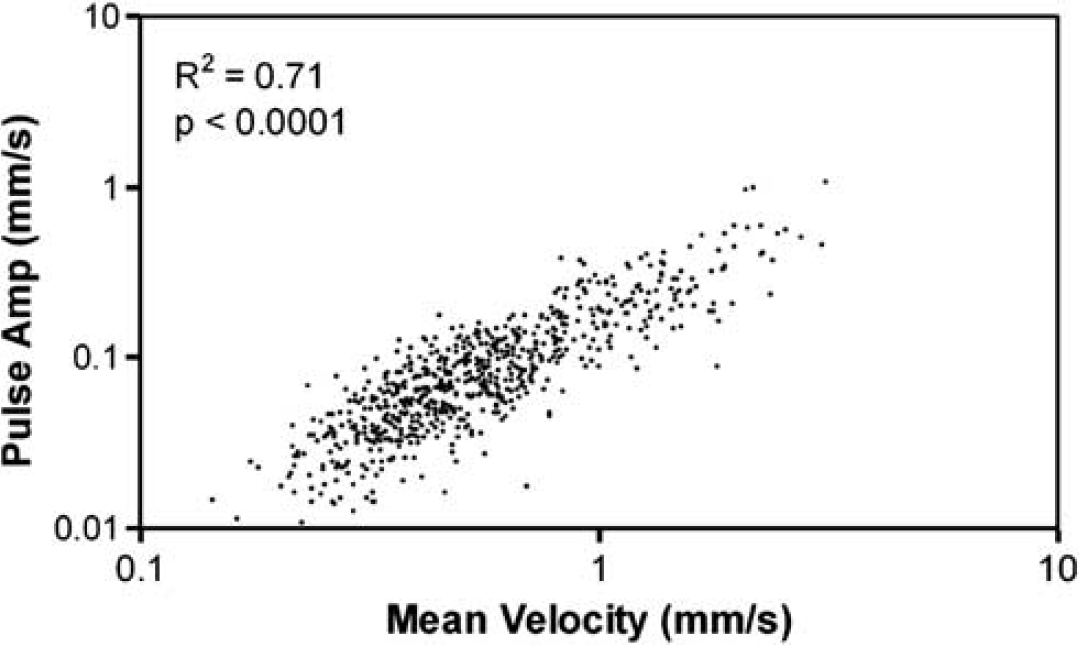
Linear relationship between absolute pulse amplitude and mean velocity plotted for all vessels and all experimental groups in the study. The highly significant, linear relationship provides the justification for using the ratio between pulse amplitude and mean velocity, the pulsatility index (PI), as the experimental measure of choice. The use of PI minimizes the variability of pulsatility measurements due to changes in absolute blood flow, which is well known to be lowered in hydrocephalus and was in fact found to be lower in the acute group.
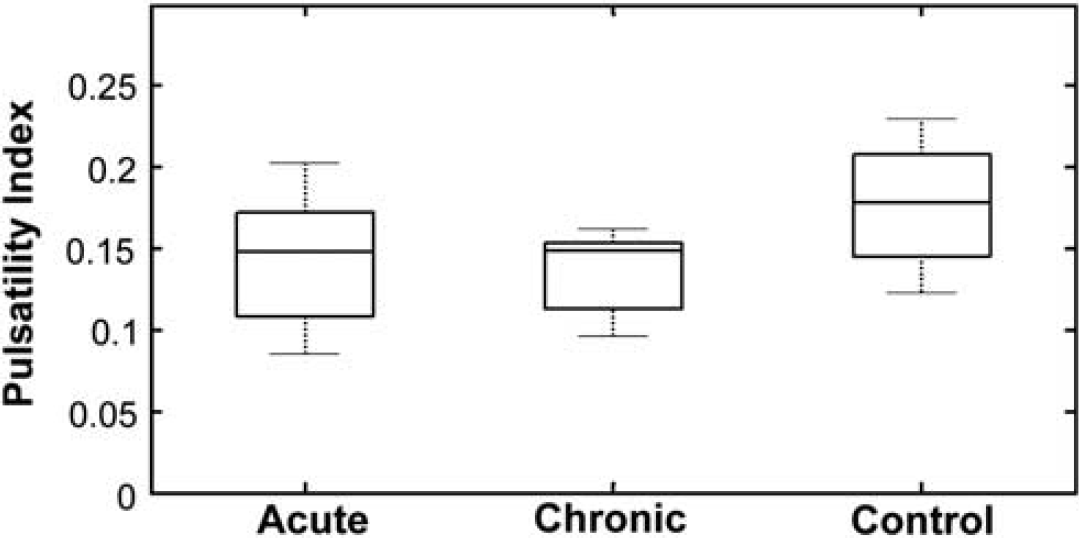
Boxplots of pulsatility index for the acute, chronic, and control groups. Without accounting for variations in vessel diameter, depth, or heart rate, there was no significant difference between the groups, other than a trend toward higher pulsatility in controls (
The second linear mixed model, which did control for these experimental parameters, showed that PI was significantly larger in controls compared with both acute and chronic animals (
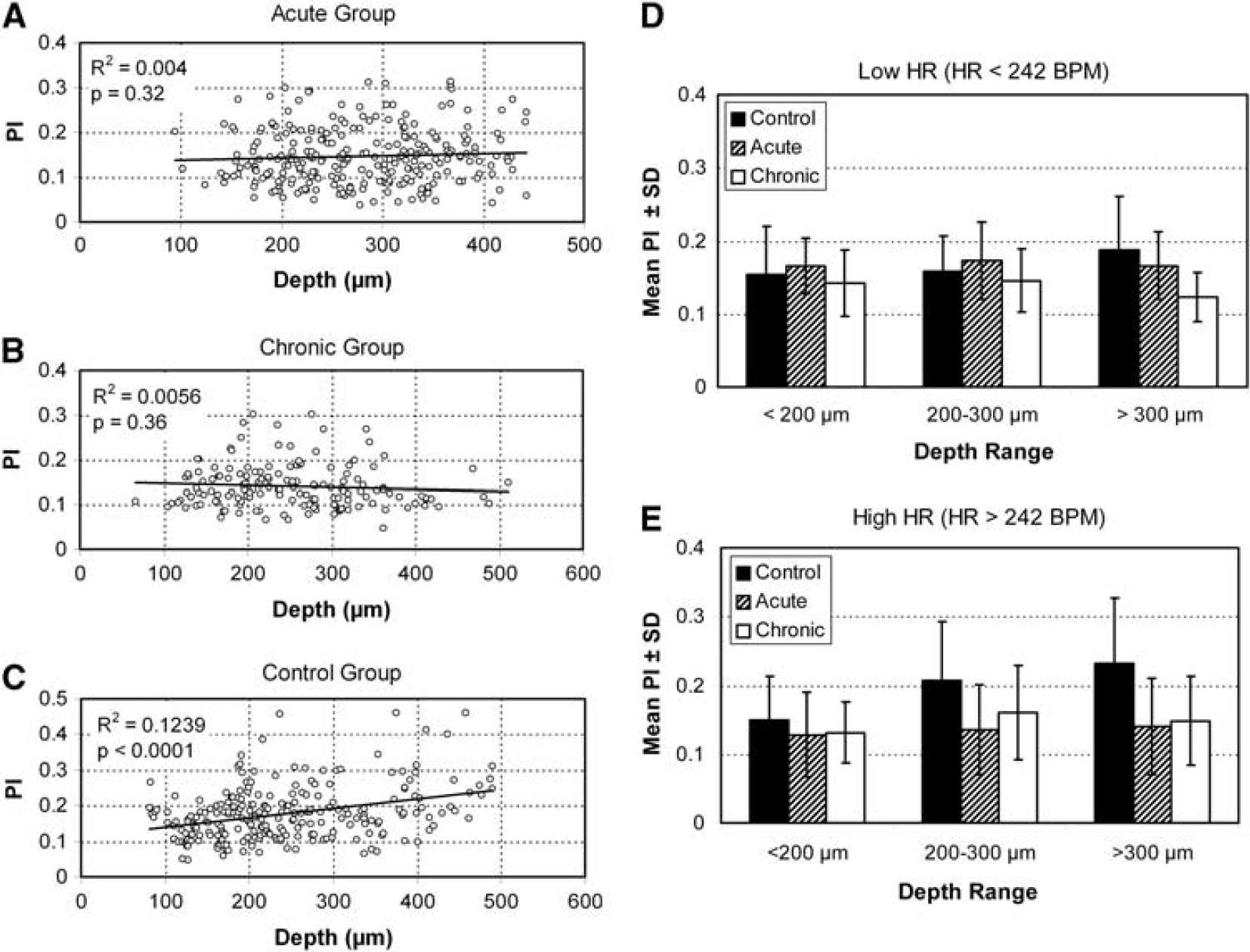
The effect of vessel depth on pulsatility index (PI) in the three experimental groups (
Pulsatility index results for the acute and chronic CH animals, averaged over all vessels in each animal, were compared with their CSF SVs and VVs (Figure 7). No correlation was found between either VV or aqueductal CSF SV and PI, for either acute or chronic CH animals. Based on prior measurements of CSF SV in intact control animals (Wagshul et al, 2009), we can conclude that there is no increase in microvascular flow pulsatility, even in the presence of a 10- to 500-fold increase in ventricular CSF pulsatility as measured at the cerebral aqueduct.
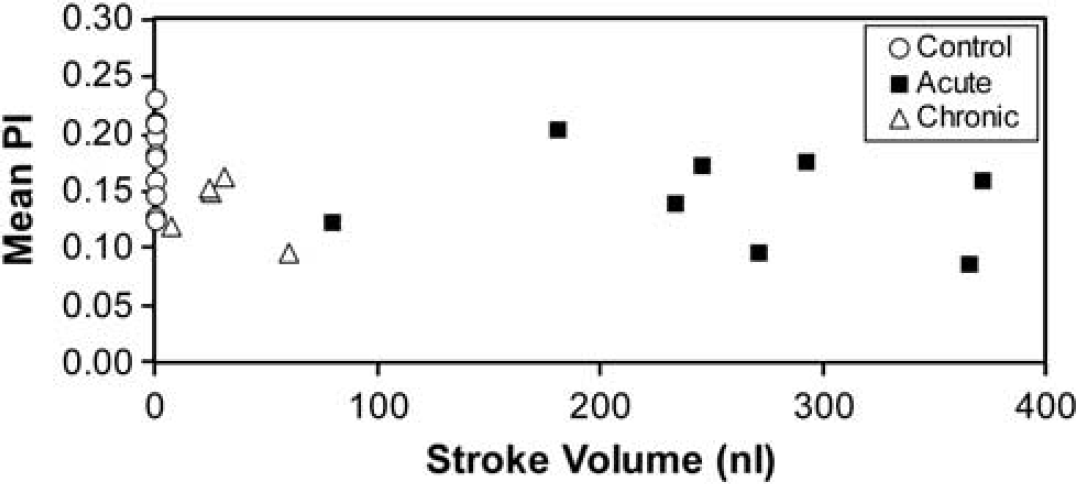
Mean pulsatility index (PI), averaged by animals, for both acute and chronic hydrocephalic animals against cerebrospinal fluid (CSF) stroke volume in the cerebral aqueduct. There was no correlation in either group. For comparison, PI is plotted for all control animals, with stroke volumes taken from the mean control stroke volume of a previous study (Wagshul et al, 2009). The plot clearly indicates that even in cases where the ventricular CSF pulsatility is markedly elevated (with a 10- to 500-fold increase compared with normal) there is no increase in cortical microvascular pulsatility.
Discussion
We have shown the feasibility of measuring flow pulsatility in the live rat neocortex from the pial surface to depths of 500 μm. We have applied the technique to measurements in a rat model of CH, although the technique is easily translatable to other small animal models and opens up the possibility of microvascular pulsatility studies in a broader context, such as stroke and atherosclerosis. Contrary to our initial hypothesis, our results indicate that pulsatility of capillary blood flow in the rat neocortex is not elevated in hydrocephalus. Statistical analyses accounting for multiple experimental factors also demonstrated a new finding: a significant increase in capillary pulsatility with depth from the pial surface in control animals that disappears in hydrocephalus.
Relationship of Pulsatility with Vessel Depth, Diameter, and Heart Rate
Three parameters were examined to assess their effect on pulsatility: depth of the vessels from the pial surface, vessel diameter, and HR. In control animals, our results show a complex dependence of pulsatility on all three parameters. Vessel diameter is expected to affect mean flow velocity, and therefore, absolute pulse amplitude. Studies documenting changes in capillary morphology also show that cortical capillary density is altered in hydrocephalus (Jones et al, 1991; Luciano et al, 2001). Such effects may give rise to changes in mean blood flow and absolute pulse amplitude in capillaries, but should not affect their ratio. Thus, to mitigate the effect of vessel diameter and density on our results, PI was chosen as the outcome measure. Our results do show a very small increase in pulsatility with vessel diameter, which may be related to changes in effective vessel compliance with size. However, there was no difference in this dependence between experimental groups, indicating that any differences in compliance between the groups that might lead to changes in pulsatility are driven by factors unrelated to vessel morphology but more likely driven by extravascular factors.
In contrast, PI showed a much stronger dependence on depth, increasing significantly with depth, in controls only. Similarly, the positive relationship between pulsatility and HR in control animals was not present in the hydrocephalic animals. Based on these depth-by-group and heart rate-by-group associations, we conducted a
While more detailed investigations will clearly be needed to fully explain these results, we can offer a few plausible explanations. The most likely candidate to explain changes in pulsations in the brain is intracranial compliance, based on the well-known pressure—volume relationship (Marmarou et al, 1975). Marmarou showed that the exponential pressure—volume relationship in the brain leads to larger pressure pulsatility with either an increase in intracranial pressure or a decrease in intracranial compliance. Given that the primary sources of intracranial compliance are the veins and CSF-filled subarachnoid spaces (because of their direct communication with the compliant thecal sac), an increase in PI with depth may simply be a result of the increased distance from these sources of compliance. Furthermore, based on these assumptions our results of an overall decrease in pulsatility with hydrocephalus would be a reflection of increased brain compliance, which while counterintuitive is in agreement with recent direct measurements of locally increased compliance in hydrocephalus using magnetic resonance elastography (Streitberger et al, 2011). It should be noted, however, that these results are in sharp contrast with findings from several other studies showing decreased compliance in hydrocephalus (Bateman, 2000; Gonzalez-Darder and Barcia-Salorio, 1989; Marmarou et al, 1975; Miyati et al, 2003). It may be that our localized capillary measurements as well as magnetic resonance elastography are effectively measures of local tissue compliance, which increases in hydrocephalus while the classic CSF infusion studies probe the global compliance of the entire craniospinal system, which decreases in hydrocephalus.
Limitations
One potential limitation of the study is related to the anesthesia delivery. Since an injectable anesthetic was used with supplemental boluses as needed rather than a continuous infusion, the level of anesthesia varied over time, and may have been responsible for some of the variability in pulsatility. In particular, studies of the effect of hypercapnia on pressure pulsatility show that the systemic arterial and CSF pulse amplitudes are elevated with hypercapnia, with the latter attributed to capillary dilation (Avezaat et al, 1980; Portnoy and Chopp, 1981). However, there is a clear correlation between anesthesia depth (and thus potentially hypercapnia, due to depressed respiratory rate) and HR. If anything, our data show increased pulsatility in controls with increased HR, not the reverse, and no change in hydrocephalic animals. A further possible concern is the effect of anesthesia on cerebral blood flow. Ketamine—xylazine has been shown to cause a decrease in cerebral flow as compared with isoflurane (Lei et al, 2001; Masamoto et al, 2010). However, this is an effect on mean blood flow and the effects of ketamine—xylazine on flow pulsatility are not known. Moreover, because all animals received the same anesthetic regimen, we do not expect the particulars of the anesthesia to have affected our final conclusions.
Making an opening in the skull may irreversibly alter intracranial dynamics, by increasing intracranial compliance, minimizing any changes in pulsatility that may have existed. However, the cranial window was immediately sealed, and was carefully observed under the surgical microscope to ensure that no air bubbles had been left within the craniectomy and that the seal was complete. In addition, experiments were performed in both acute and chronic hydrocephalus animals, which presumably have different levels of elevated intracranial pressure (anecdotally, there was slightly more brain herniation through the craniectomy in acute animals) and no difference was found between these groups. Nonetheless, our null results were consistent in both groups of animals, implying that changes in intracranial dynamics from making the cranial window did not affect the results.
Ventriculomegaly was less pronounced in the chronic CH group compared with the acute CH group. The VVs of these animals clearly fit our criteria for hydrocephalus, with enlarged ventricles as well as elevated CSF SV compared with controls. Ventricular dilation in this hydrocephalus model, as in many other hydrocephalus models, generally varies over a wide range, and we have to consider it an unfortunate, unexplained coincidence that all animals in the chronic CH group were only mildly hydrocephalic. We have shown in a prior study, as well as our extensive unpublished experience with this model, that chronic animals can experience the entire range of mild-to-severe ventricular dilation (Wagshul et al, 2009). Nonetheless, even with the limited ventriculomegaly in this group, we were able to show that hydrocephalus leads to reduced capillary pulsatility compared with controls at the deeper cortical levels.
Differentiating Flow and Pressure Pulsatility
To understand the implications of our results, and how they might relate to other measurements of pulsatility in the literature, a brief explanation is in order regarding the important difference between cardiac-induced pulsations in pressure waves as opposed to pulsations in flow velocity within CSF or vascular spaces; for a complete overview, the reader is referred to a recent review article on the topic (Wagshul et al, 2011). Briefly, all cardiac-induced pulsations in the brain can be assumed to originate from variations in pressure and flow in the carotid and basilar arteries feeding into the brain. In hydrocephalus, studies have shown changes in both intracranial pressure pulsatility (Carrera et al, 2010; Eide et al, 2007; Eide and Sorteberg, 2008; Eide and Saehle, 2010; Hu et al, 2008) and flow pulsatility (Bakker et al, 2002; Baledent et al, 2001; Bateman et al, 2005; Kahlon et al, 2007; Leliefeld et al, 2009; Miyati et al, 2003; Nadvi et al, 1995). However, a critical distinction between pressure and flow effects is that pressure waves are transmitted very rapidly throughout the brain, so that pressure pulsations within the ventricle, for example, are nearly identical to those measured within the parenchyma. Flow, however, relies on the physical transfer of fluid from one compartment to another, and therefore marked differences in flow velocities, and in flow pulsatility, can exist from one part of the brain to another. Therefore, one cannot necessarily reach any conclusions with respect to flow pulsatility based on the pressure pulsatility literature or vice versa. For example, even in the presence of elevated pressure pulsations throughout the brain, flow pulsations may be normal in certain compartments, since flow pulsations depend on the existence of a pressure pulsation gradient across the flow compartment (e.g., from the arterial to the venous side of a capillary bed). Thus, the discussions below are restricted to observations of flow pulsations in hydrocephalus alone.
Significance of Pulsatility in Communicating Hydrocephalus
The primary motivation for studying capillary pulsatility in hydrocephalus was to elucidate a possible role of microvascular pulsations in the pathogenesis of hydrocephalus. While ventriculomegaly is a well-explained phenomenon in cases with a clear obstruction to ventricular outflow, in CH clear-cut explanations for the mechanisms of ventricular dilation are lacking (Egnor et al, 2002; Greitz, 2004). A number of early studies suggested that ventricular dilation in CH may be a result of elevated ventricular pulsations, and that reduction of the CSF pulse wave can inhibit ventricular dilation (Bering, 1962; Wilson and Bertan, 1967). The most striking evidence came from a study showing that ventricular dilation could be induced in the absence of any CSF obstruction by artificially elevating the CSF pulse wave with a pulsating, intraventricular balloon (Pettorossi et al, 1978). Finally, elevated pulsations have also been extensively reported in clinical studies (Bradley et al, 1996; Greitz et al, 1994; Luetmer et al, 2002; Miyati et al, 2003). While there have been some attempts to use measures of CSF pulsatility to guide shunt surgery (Bradley et al, 1996), a number of recent trials have shown this to be an inadequate indicator of shunt success (Algin et al, 2010; Dixon et al, 2002; Kahlon et al, 2007).
To understand the connection between ventricular CSF pulsations and pulsatility at the capillary level, it is important to realize that almost all pulsatility in the brain is cardiac in nature and originates from the arterial vasculature. However, the route of pulsations from the arteries into the ventricles is still debated (Bering, 1962; Du Boulay, 1966; Egnor et al, 2002; Greitz, 2004). Since no studies have shown increased
This pulsatility redistribution theory posits that arterial pulsations entering the brain are normally dampened via the Windkessel mechanism (Fung, 1997) and coupled into subarachnoid CSF, extracerebral veins, and venous sinuses. This gives rise to extracerebral venous pulsatility and pulsatile CSF flow in the SAS, which has been measured by magnetic resonance imaging at the foramen magnum (Alperin et al, 2000; Quigley et al, 2004). In CH, decreased global intracranial compliance may disrupt the normal intracranial flow dynamics, diminishing the transfer of arterial pulsations into subarachnoid CSF and veins and producing a decrease in CSF pulsatility at the foramen magnum (Greitz, 1993). In particular, in our model of basal cistern obstruction mimicking clinical cases such as subarachnoid hemorrhage and meningitis that also block this pathway, one would expect a decrease in pulsations transferred through the basal cisterns into the spinal SAS. With the removal of this major pulsatility dissipation pathway, arterial pulsations can propagate undampened into the intracranial microvasculature, which has recently been shown to affect normal hemodynamics (Silacci et al, 2001; Ziegler et al, 1998). Furthermore, increased pulsatility of the entire intracranial microvasculature would be expected to result in increased brain pulsatility, which acting on the walls of the lateral ventricles would produce the well-documented increased ventricular CSF pulsatility. So, this theory predicts that undampened arterial pulsations will be transmitted into the microvasculature, and should be detectable as increased flow pulsation in cerebral capillaries.
The present study was thus designed to detect increased flow pulsations in neocortical capillaries with the induction of CH as a test of a pulsatility redistribution theory. We have shown that, at least on initial analysis, this theory is not correct: neocortical capillaries do not exhibit increased pulsatility with hydrocephalus induction. On the contrary, we have shown a significant decrease in capillary pulsatility with hydrocephalus, and have argued that these changes may be a product of the compliance changes in the hydrocephalic brain.
Alternate Explanations for Low Pulsatility
Based on our findings, we would conclude that (1) the redistribution theory is not an accurate representation of intracranial flow dynamics in hydrocephalus, or (2) pulsatility redistribution only causes local changes in pulsatility, such as in the choroid plexus or periventricular tissue, or (3) there is no relationship between macroscopic CSF flow pulsations and microscopic vascular pulsation. If increased capillary pulsatility is only manifest deep within the periventricular regions, and is not apparent in the superficial neocortical layers, our technique would not be capable of showing such changes. Our maximum imaging depth was 0.5 mm, while the depth of the periventricular white matter is ∼1.5 mm in our most severely hydrocephalic rats. The theoretical maximum imaging depth for multiphoton microscopy is ∼1 mm without resorting to much more invasive surgical procedures; recent advances have allowed
Conclusion
This study is the first attempt to measure capillary pulsatility deep in the rat neocortex. We have shown that capillary pulsatility normally increases with increasing depth from the pial surface. Our attempt to provide supportive evidence for a pulsatility redistribution theory of CH showed that if such redistribution does occur either it is not evident as a global increase in flow pulsatility throughout the cranium or it is not evident in the later stages of hydrocephalus when ventricular size has stabilized. Furthermore, we found that the normal increase in pulsatility with depth disappears with hydrocephalus induction, and explanations of this effect based on local intracranial compliance changes have been suggested. Future studies will investigate capillary pulsatility at earlier time points, potentially within hours of induction, when the kaolin blockage may cause changes in pulsation distributions but ventricular enlargement has not yet occurred.
Footnotes
Acknowledgements
The authors thank Dr Jie Li of SUNY Upstate Medical University for assistance with the surgical procedures, Dr Joseph Madsen of Harvard Medical School for help with the initial design of the experiments, and Dr Noam Alperin of the University of Miami for valuable discussions regarding the manuscript and interpretation of our results. The authors thank the Brain Child Foundation and STARS-kids Foundation for financial support of our work.
The authors declare no conflict of interest.