
Abstract
Epidemiologic studies have shown that foods rich in polyphenols, such as flavanols, can lower the risk of ischemic heart disease; however, the mechanism of protection has not been clearly established. In this study, we investigated whether epicatechin (EC), a flavanol in cocoa and tea, is protective against brain ischemic damage in mice. Wild-type mice pretreated orally with 5, 15, or 30 mg/kg EC before middle cerebral artery occlusion (MCAO) had significantly smaller brain infarcts and decreased neurologic deficit scores (NDS) than did the vehicle-treated group. Mice that were posttreated with 30 mg/kg of EC at 3.5 hours after MCAO also had significantly smaller brain infarcts and decreased NDS. Similarly, WT mice pretreated with 30 mg/kg of EC and subjected to
Introduction
Numerous epidemiologic studies have revealed a strong inverse correlation between ischemic heart disease and consumption of red wine and certain fruits and vegetables that contain high levels of flavonoids and other polyphenols (Simonyi et al, 2005). Flavanols (e.g., epicatechin (EC) and catechin) and their monomers/oligomers/polymers, known as procyanidins, compose a major category of secondary polyphenolic plant metabolites. Mounting evidence suggests a protective role for various polyphenols and flavonoids in cerebral ischemia (Lee et al, 2004; Lee et al, 2000; Shah et al, 2005; Shin et al, 2006; Simonyi et al, 2005). It is noteworthy also that flavanols have been shown to improve vasorelaxation (Duarte et al, 1993; Karim et al, 2000). The simple chemical structure of flavanols may interact with specific cellular and molecular targets, thereby mediating a wide range of biologic activities.
Studies have revealed that induction of phase II antioxidant enzymes through transcriptional activation is mediated through transcriptional factor Nrf2 and antioxidant-response elements (AREs). In these paradigms, polyphenols or electrophylic agents can target a specific set of genes that encode phase II enzymes, which include heme oxygenase 1 (HO1), nicotinamide adenine dinucleotide phosphate (NADPH) quinone oxidoreductase 1, and γ-glutamyl cystein ligase. These enzymes provide protection by regulating and maintaining intracellular redox states (Gong et al, 2002; Itoh et al, 2004). Of these, HO1 has been reported to have the most AREs on its promoter, making it a highly effective therapeutic target for protection against neurodegenerative diseases. It offers protection in part by degrading its pro-oxidant substrate, heme, and generating the antioxidants biliverdin and bilirubin (Doré et al, 1999). The reaction also releases one molecule of iron from the core of the heme porphyrin rings. This iron may then increase the cellular levels of ferritin, which can prevent additional generation of free radicals. Carbon monoxide (CO), another by-product of this reaction, may also have vasodilatory actions along with reported antiapoptotic and antiinflammatory properties at low concentrations; we have recently shown it to offer beneficial effects in transient ischemia (Zeynalov and Doré, 2009).
Increasing evidence suggests that HO1 can be upregulated through the Nrf2/ARE-mediated pathway (Satoh et al, 2006; Zhao et al, 2006). We have shown that Nrf2, a key regulator of the HO1 gene, has an important role in protection against cerebral ischemic stroke (Shah et al, 2007). Considering the beneficial properties of polyphenols and the possible role of the Nrf2/HO1 pathway, in this study we used
Materials and methods
Animals
All animal protocols were approved by the institutional animal care and use committee of Johns Hopkins University. HO1 knockout (HO1−/−) mice were originally generated by Drs Poss and Tonegawa (Poss and Tonegawa, 1997). Mouse genotype was assessed by PCR and additionally confirmed by standard Western blot analysis. Male HO1−/−, Nrf2−/−, and wild-type (WT) mice (20 to 25 g; 7 to 8 weeks old) had access to food and water ad libitum and were housed under controlled conditions (23°C ± 2°C; 12-h light/dark periods). Animals were provided Teklad Global 18% Protein Rodent Diet (Harlan Holding, Inc., Wilmington, DE, USA), formula 2018S, which is a fixed-formula, autoclavable pellet chow that contains no nitrosamines and a low level of natural phytoestrogens, with 18% protein (non-animal) and 5% fat for consistent growth, gestation, and lactation. All mice were randomly assigned to the different experimental groups.
Gavage Administration of Epicatechin
Epicatechin was administered orally (per kilogram of body weight) by gavage with efforts made to minimize stress to the mice. For pretreatment studies, one dose of EC (2.5, 5, 15, or 30 mg/kg) or distilled water (control) was administered 90 minutes before MCAO; in posttreatment experiments, 30 mg/kg EC or distilled water was administered at 3.5 or 6 hours after MCAO. We selected these doses based on previous findings in which a 30-mg daily dose resulted in 7.3 ng/mg tissue (wet weight) (−)EC and 16.0 ng/mg tissue (wet weight) 3′-
Induction of Transient MCAO and Measurement of Infarct Size, Neurologic Deficits, Blood Gases, and Physiologic Parameters
Mice were subjected to MCAO as described previously (Shah et al, 2006). In brief, mice were anesthetized with halothane (3% initial, 1% to 1.5% maintenance) in O2 and air (80%:20%). Relative cerebral blood flow (CBF) was measured by laser-Doppler flowmetry (DRT4; Moor Instruments Ltd, Devon, UK) through a microfiber affixed to the skull over the area of parietal cortex approximately 6 mm lateral and 1 mm posterior of the bregma. Through a midline incision in the neck, a silicone-coated 7–0 Ethilon nylon filament (Ethicon, Inc., Somerville, NJ, USA) was advanced into the internal carotid artery through the severed external carotid artery to block the blood circulation to the middle cerebral artery, or Circle of Willis. A drop in CBF of ⩾80% was considered to be a successful occlusion. Mice not attaining the required decrease in CBF were excluded from the study. Cortical perfusion values were expressed as a percentage relative to baseline. Mice were moved to a 32°C humidity/temperature-controlled chamber to maintain a body temperature of 37°C during the 90-minute occlusion. With the mice anesthetized, reperfusion was initiated by withdrawing the filament. Mice were returned to the humidity/temperature-controlled chamber for 2 hours before being returned to their respective cages. The stroke was considered successful if no subarachnoid hemorrhage was observed, a lesion was produced, and the mouse survived to the required end point. To evaluate motor deficits, sensorimotor performance was evaluated on a 4-point neurologic deficit severity scale as described previously (Shah et al, 2006). Mice were assessed for neurologic deficits at 24 hours (pretreatment group) or 72 hours (posttreatment group) after occlusion by the following scale: 1, no deficit; 2, forelimb weakness; 3, inability to bear weight on the affected side; and 4, no spontaneous motor activity. After being tested, the mice were anesthetized, and their brains were removed and cut into 2-mm coronal sections, which were stained with 2,3,5-triphenyltetrazolium chloride (TTC; Sigma, St Louis, MO, USA). The sections were scanned individually by a video imaging system and analyzed using image analysis software (SigmaScan pro 4 and 5; Systat, Inc., Point Richmond, CA, USA). Measurements of pH, PaO2, PaCO2, and CBF were made in a separate cohort of mice.
Induction of NMDA-Induced Acute Excitotoxicity and Quantification of the Lesion Volume
Mice were administered a single oral dose of either saline or 30 mg/kg EC 90 minutes before unilateral intrastriatal NMDA injection. After body weight and rectal temperature were recorded, mice were anesthetized and placed on a stereotaxic stand. Then, 15 nmol of NMDA were injected slowly into the right striatum. The injection needle was slowly withdrawn, the hole was blocked with bone wax, and the skin was sutured. On recovery from anesthesia in a thermoregulated chamber, mice were transferred to their home cages. Throughout the experimental procedure, the rectal temperature of each mouse was monitored and maintained at 37.0°C ± 0.5°C. After 48 hours, mice were deeply anesthetized and transcardially perfused and fixed. Brains were equilibrated in 30% sucrose, frozen, and cut sequentially into 25-mm sections on a cryostat. The sections were stained with Cresyl Violet to estimate the lesion volume (Ahmad et al, 2006).
Neuronal Cell Cultures, Cell Survival, and Caspase Assays
Embryonic cortical neuronal cells were isolated from 17-day embryos of timed pregnant mice, and postnatal cortical neurons were obtained from 1- to 2-day-old mice, as previously detailed (Shah et al, 2007). Neurons (5 × 105 cells/well) were plated in serum-free Neurobasal medium supplemented with 1 mmol/L Glutamax (Invitrogen, Carlsbad, CA, USA) and B27 supplement onto 24-well plates coated with poly-D-lysine. Cells were maintained at 37°C in a 95% air and 5% CO2 humidified atmosphere. All experiments were performed after 14 days
Neuronal survival was assessed with the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) colorimetric assay, as described (Shah et al, 2007). Cell viability was also measured by the lactate dehydrogenase (LDH) assay, which assesses the membrane integrity of cells. After experimental treatment, the culture medium was collected and mixed with substrate, enzyme, and dye solution. After 30 minutes of incubation in the dark, the reaction was terminated by adding 1:10 volume of 1N hydrochloric acid. Absorbance was measured at 490 nm.
The caspase 3/7 activity of neuronal samples was measured according to the manufacturer's instructions (Promega, Madison, WI, USA). Homogeneous caspase reagent was added to the cells, which were then incubated at room temperature for 18 hours in the dark. The fluorescence of each sample was measured at excitation and emission wavelengths of 485 and 530 nm, respectively. Experiments were repeated with at least three separate batches of cultures.
Western Blot Analysis
Cytosolic and nuclear fractions were isolated from the cortical neurons as described previously (Shah et al, 2007). Protein concentration was determined by a BCA kit (Pierce, Rockford, IL, USA). Equivalent amounts of protein per sample were resolved through sodium dodecyl sulfate-polyacrylamide gel electrophoresis on 10% gels. The proteins were electrophoretically transferred to a nitrocellulose membrane, which was blocked for 1 hour at 22°C with 5% dried milk and then exposed to the primary antibody overnight at 4°C and to the secondary antibody in 5% dried milk for 1 hour at 22°C. Immunocomplexes were visualized by enhanced chemiluminescence detection (ECL; Amersham, Piscataway, NJ, USA). Each Western blot shown is a representative of at least three separate experiments.
Immunocytofluorescence Staining
After being treated with EC, neurons were permeabilized for 2 minutes with 0.5% Triton X-100 and then fixed with 3% paraformaldehyde for 20 minutes. Cells were first incubated with normal goat serum to block nonspecific binding and then with primary antibodies to NeuN (a neuronal marker; Chemicon, Temecula, CA, USA) or Nrf2 (Sigma) for 30 minutes. Cells were washed and then incubated with rhodamine-conjugated, affinity-purified donkey anti-rat IgG (H + L) and fluorescein isothiocyanate (FITC)-conjugated, affinity-purified goat anti-rabbit IgG (H + L) (Jackson ImmunoResearch Laboratories, West Grove, PA, USA) secondary antibodies for 30 minutes. Images were captured with a CoolSNAP HQ camera (Image Processing Solutions, Inc., North Reading, MA, USA) using OpenLab software (Improvision Inc., Boston, MA, USA).
Statistical Analysis
Data, expressed as mean ± s.e.m., were analyzed using Student's
Results
Protective Effect of Epicatechin Pretreatment in Transient MCAO
Pretreatment of WT mice with EC 90 minutes before MCAO significantly and dose-dependently protected against neurologic deficit and brain injury. Infarct volumes of mice pretreated with EC were significantly smaller than those of vehicle-treated mice. Infarct volumes were 40.0% ± 3.2% (
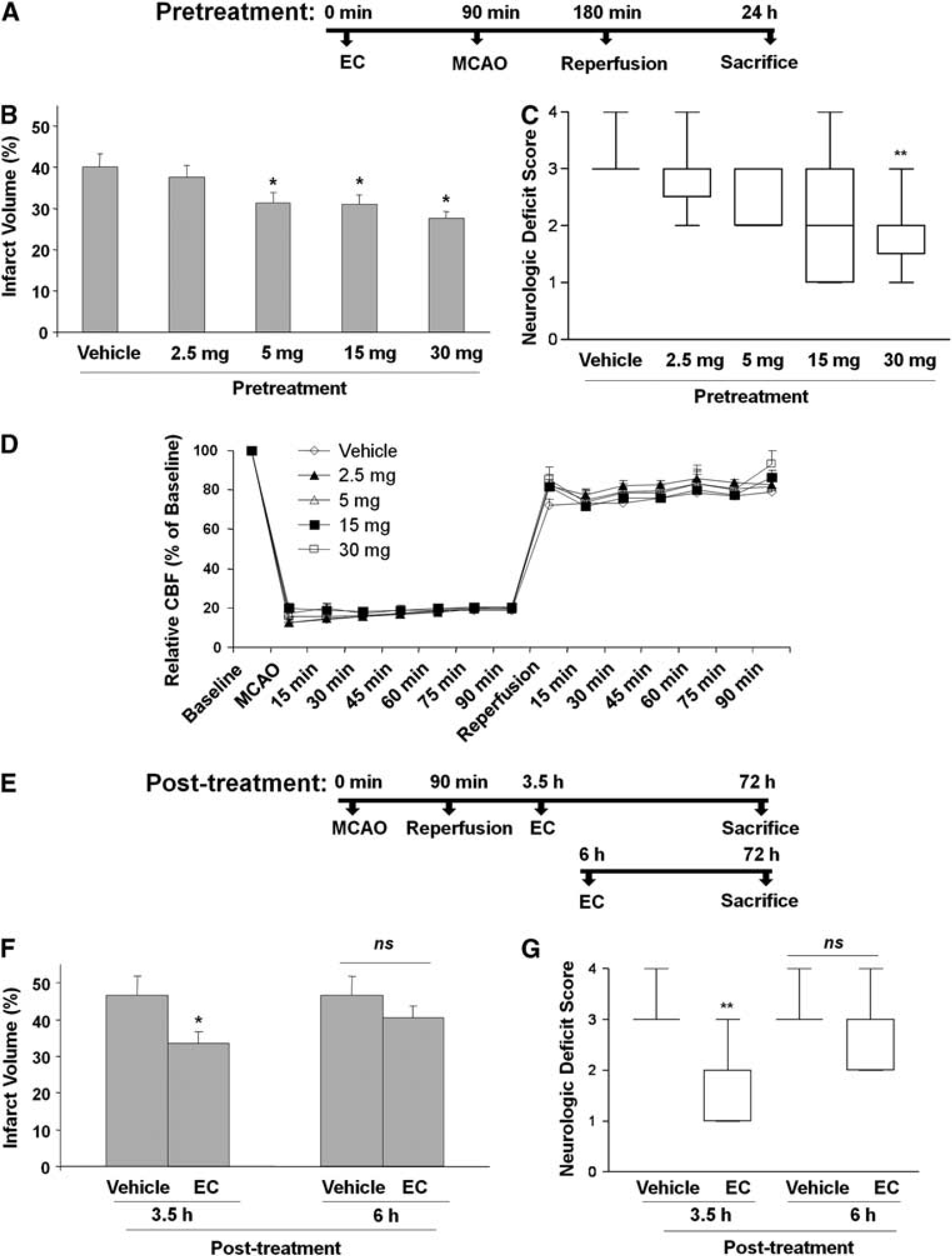
(−)-Epicatechin (EC) is protective against transient middle cerebral artery occlusion (MCAO)-induced cerebral damage in wild-type (WT) mice. (
Physiologic Parameters and Cerebral Blood Flow
In a separate cohort of mice also pretreated with vehicle or EC (2.5, 5, 15, and 30 mg/kg;
Protective Effect of Epicatechin Posttreatment in Transient MCAO
In mice posttreated with 30 mg/kg EC, no mortality was observed, but three mice in the vehicle-treated group died after 48 hours. The cause of the deaths appeared to be severe edema, and not a direct result of the surgical procedure. Mice that were posttreated with EC 3.5 hours after MCAO had significantly smaller infarct volumes (33.5% ± 3.0%;
Protective Effect of Epicatechin Pretreatment in NMDA-Induced Acute Excitotoxicity
Unilateral injection with 15 nmol of NMDA produced brain damage in the mouse ipsilateral striatum (Figure 2B, left) that was significantly attenuated by pretreatment with 30 mg/kg EC (Figure 2B, right). At 48 hours, lesion volume of the EC-treated group (6.8% ± 1.0%;
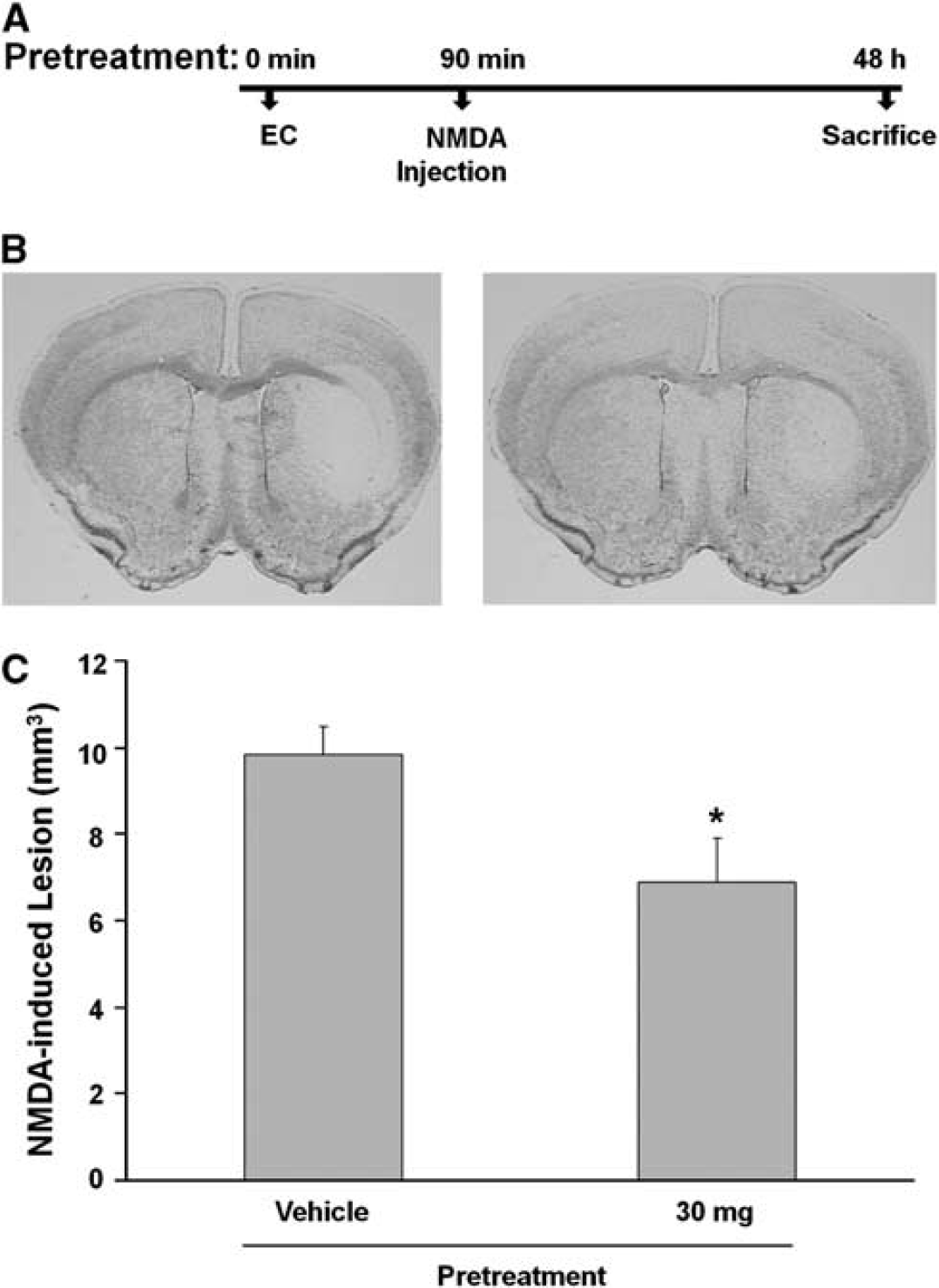
Epicatechin (EC) treatment reduces
Neuroprotection of Epicatechin Against Oxidative Stress in Neuronal Cultures
To study the EC-mediated neuroprotection
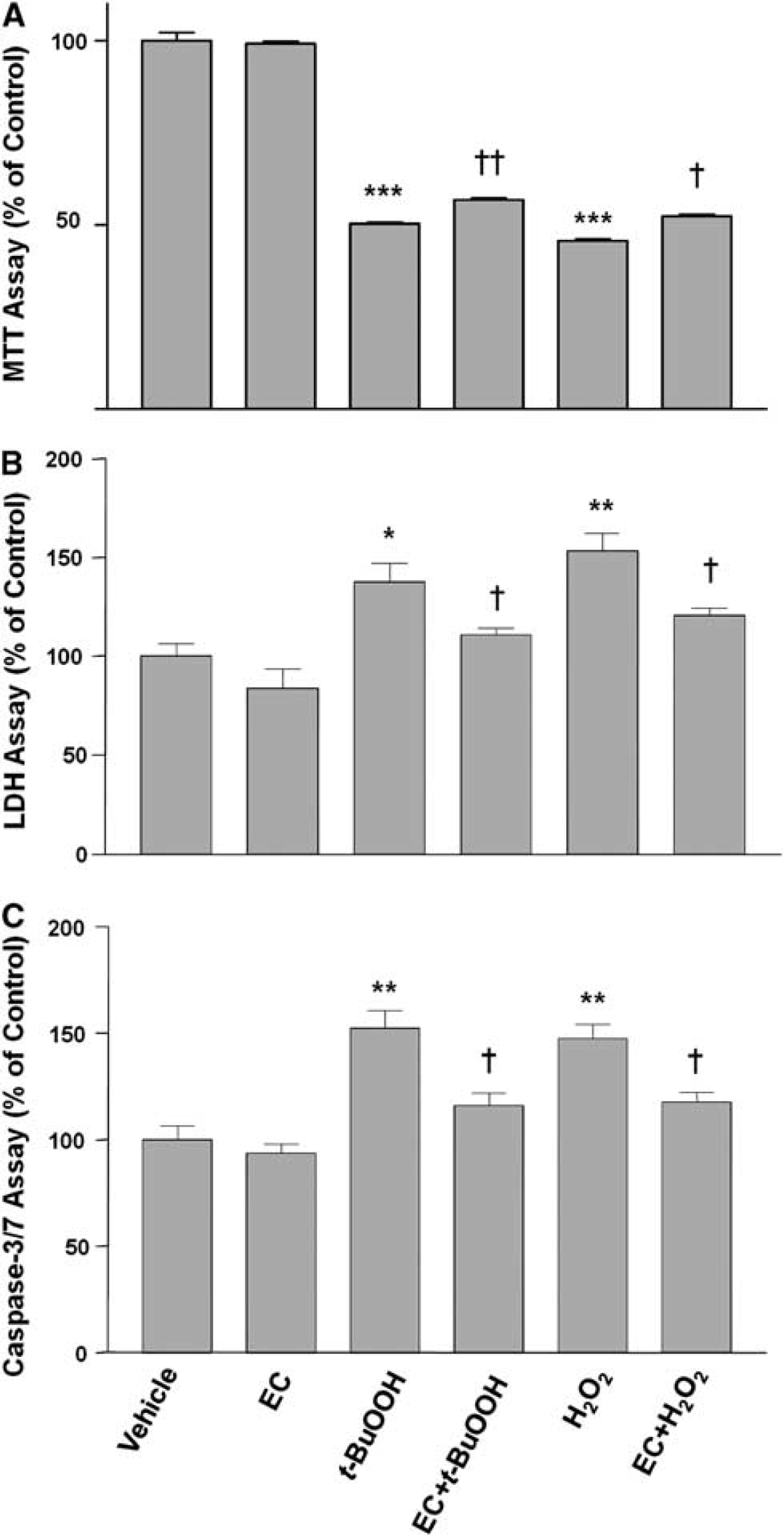
Neuronal protection by epicatechin (EC). Neurons were grown for 24 hours in culture medium alone (vehicle) or in the presence of
Induction of HO1 Expression and Nrf2 Translocation by Epicatechin in Neuronal Cultures
To analyze whether EC can induce HO1 protein expression, primary neuronal cultures were exposed to increasing concentrations of EC (0.1 to 100 μmol/L). Western blot analysis showed that EC increased HO1 protein expression in a dose-dependent manner (Figure 4A). Even at the highest concentration tested, 100 μmol/L, EC did not affect cell survival. Therefore, we used 100 μmol/L EC for the remainder of the
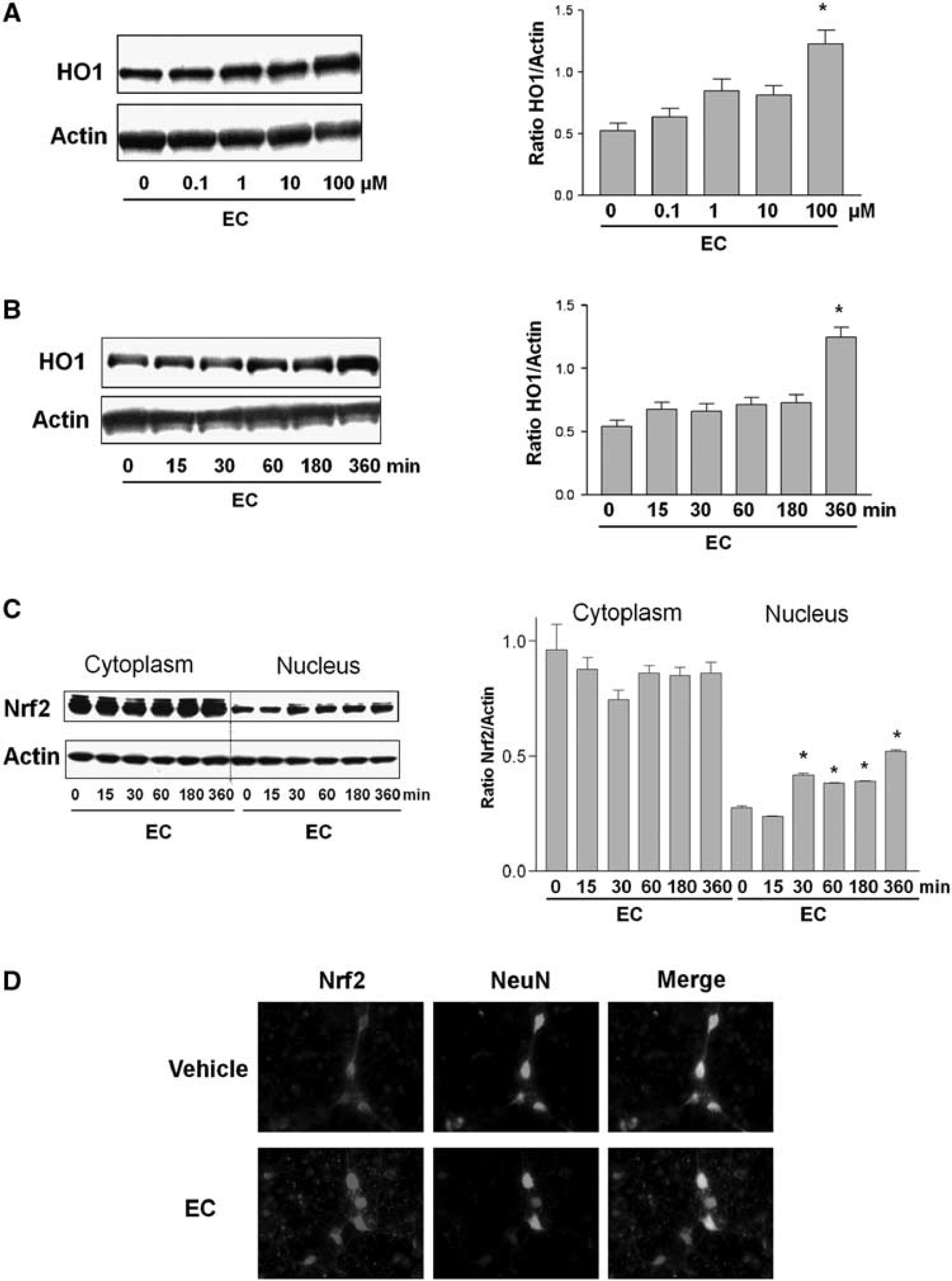
Effects of epicatechin (EC) on heme oxygenase 1 (HO1) and Nrf2 protein expression and nuclear accumulation in cortical neurons. (
The protein expression of HO1 also increased with increased exposure time to EC (Figure 4B). If the transcriptional factor Nrf2 is partially responsible for the upregulation of HO1 by EC, one would expect Nrf2 to accumulate in the nucleus of cells as a result of EC exposure. To analyze whether this occurs, primary cultured cortical neurons were exposed to EC (100 μmol/L), and cytoplasmic and nuclear fractions were analyzed by Western blot. The presence of Nrf2 in the nuclear fraction increased in a time-dependent manner. It increased rapidly in the first 30 minutes and remained elevated throughout the full 6-hour time course (Figure 4C). The protein samples were also probed with Lamin B (a nuclear marker) to verify the enrichment of the nuclear extracts. On Western blots, bands were clearly observed in the nuclear extract but were undetectable in the cytoplasmic extract (data not shown).
Next, we used indirect immunofluorescence microscopy to show that EC regulates Nrf2 distribution in neurons. In the control group, Nrf2 (red fluorescence) was detected in the neuronal cell body and dendritic trees and axons (Figure 4D). The nuclear marker 4,6-diamidino-2-phenylindole (DAPI; blue fluorescence) was used to determine the nuclear area (data not shown). The neuronal marker NeuN (green fluorescence) was detected mainly in soma and to a lesser extent in the axon–dendritic compartment. The merged image revealed colocalization of Nrf2 with NeuN. After the neurons had been exposed to EC for 30 minutes, Nrf2 appeared to be concentrated within the nucleus, with less signal remaining in the cytoplasm or axon–dendritic compartments.
HO1 Is at Least Partially Required for the Neuroprotective Effects of Epicatechin in vivo and In Vitro
To test whether HO1 might mediate some of the protection associated with EC, EC was administered to HO1−/− mice to determine whether protection against transient ischemia-induced brain injury was still apparent. HO1−/− mice pretreated with 30 mg/kg EC 90 minutes before MCAO developed infarcts (37.1% ± 3.0%;
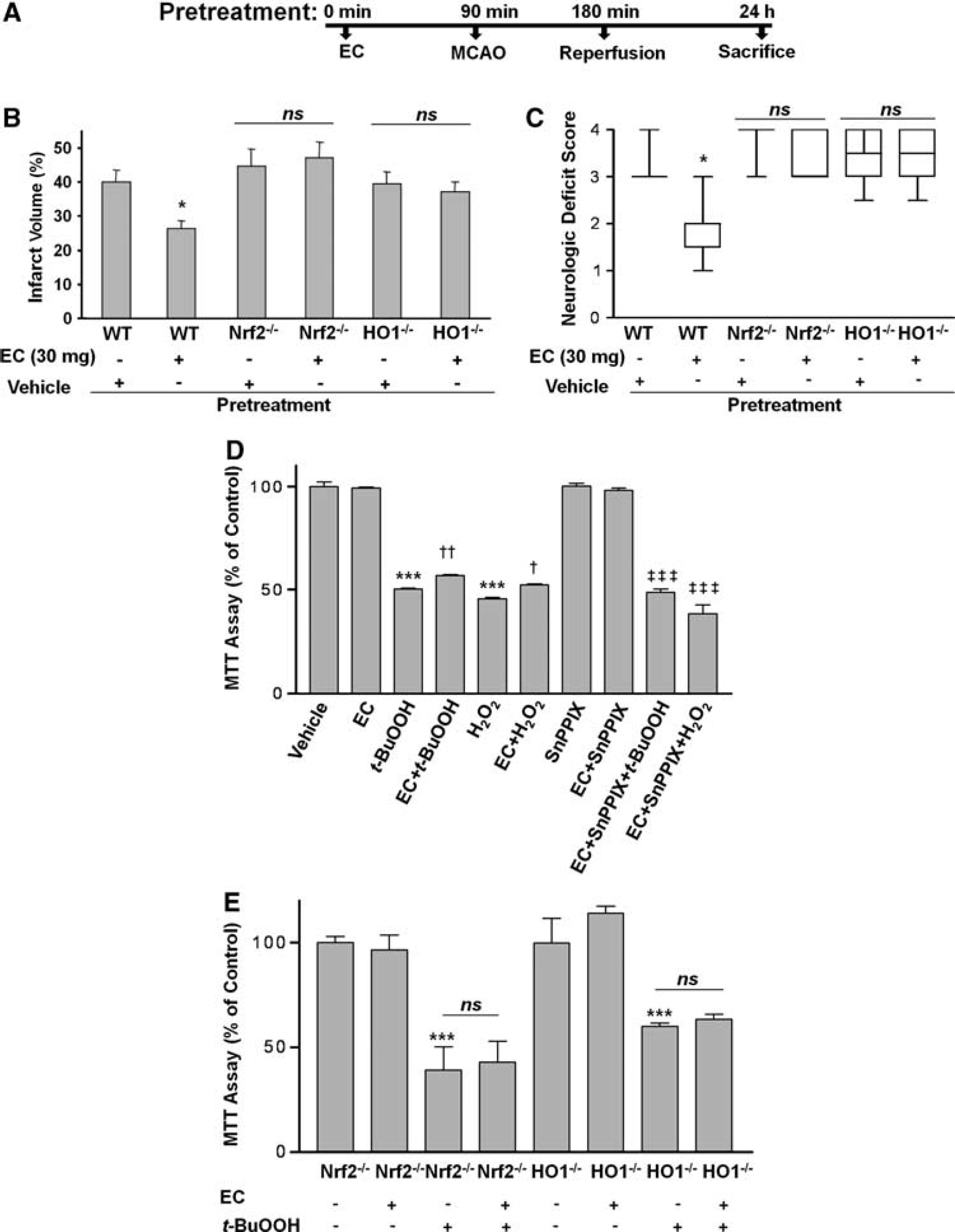
The protective effect of epicatechin (EC) is lost in Nrf2−/− and HO1−/− mice. (
To further support the concept that HO1 is needed for EC protection of neuronal cells during oxidative stress caused by H2O2 or
Nrf2 Is Required for the Neuroprotective Effect of Epicatechin in vivo and In Vitro
To better elucidate the mechanism by which EC effects neuroprotection and further substantiate the pathway of HO1 regulation, we investigated the events upstream. Evidence in the literature suggests that HO1 is upregulated through the Nrf2 cascade (Satoh et al, 2006; Shih et al, 2005). To determine whether EC works through this interdependent pathway, we pretreated Nrf2−/− mice with vehicle or 30 mg/kg EC and then subjected them to the 90-minute MCAO protocol. After 24 hours of reperfusion, mice were evaluated for NDS and then killed. Nrf2−/− mice pretreated with 30 mg/kg EC 90 minutes before MCAO developed infarcts (40.1% ± 4.5%;
Discussion
In this study, we tested the hypothesis that EC is neuroprotective against brain injury induced by transient MCAO or NMDA through the upregulation of HO1 and Nrf2. We found that EC protects the brain from transient ischemia-induced injury when administered 90 minutes before or 3.5 hours after MCAO, a finding that correlated with significant functional recovery after the onset of ischemia. The acute excitotoxicity induced by NMDA was also attenuated in mice pretreated with EC. However, treatment of HO1−/− and Nrf2−/− mice with EC failed to protect against the damage induced by brain ischemia. Our
Several studies have revealed that a therapeutic window of approximately 6 hours exists between the onset of ischemia and irreversible neuronal death (Williams et al, 2004, 2003; Xu et al, 2006). Therefore, it is desirable that neuroprotective interventions should be attempted before a stroke occurs or very soon afterward. Interventions that have the potential to reverse the cascades of injury should target mechanisms underlying delayed ischemia-induced neuronal injury, especially in the penumbra region. We followed the guidelines set by the Stroke Therapy Academic Industry Roundtable (STAIR) for stroke preclinical studies (Stroke Therapy Academic Industry Roundtable, 2001), which include assessing functional outcomes, therapeutic window, and long-term effects. We found that EC significantly and dose-dependently improved the outcome after MCAO when administered before ischemia or 3.5 hours after ischemia, but 30 mg/kg administered 6 hours after MCAO offered no significant protection, suggesting that acute posttreatment is required for significant therapeutic efficacy. Another criterion of STAIR is that the drug must cross the blood–brain barrier. Available evidence indicates that EC does cross the blood–brain barrier (Abd El Mohsen et al, 2002), as epicatechin glucuronide and 3′-
The literature provides evidence that EC is protective in many disease states (Natsume et al, 2004). For example, epidemiologic data have shown that in Kuna Indians, chronic consumption of an EC-rich cocoa drink has cardiovascular benefits that result from augmented levels of nitric oxide (Schroeter et al, 2006). In addition, it has been reported that EC consumption enhances cognition and spatial memory by increasing angiogenesis and neuronal spine density in the dentate gyrus of the hippocampus (van Praag et al, 2007). Our current results reveal that HO1 is at least partially required for EC activity. A number of cerebral ischemia studies indicate that neuroprotective agents exert an effect through the Nrf2 detoxifying pathway (Satoh et al, 2006; Shih et al, 2005; Zhao et al, 2006). This pathway has also been suggested to be at least partially mediated through HO1 induction by electrophiles (Gong et al, 2002; Itoh et al, 2004; Loboda et al, 2008). Our finding that Nrf2−/− mice were not protected against ischemic damage provides additional evidence that the protective effect of EC is mediated by activating the Nrf2/HO1 pathway. The data support our previous study, in which we showed that HO1 protects against NMDA-induced acute brain damage in mice (Ahmad et al, 2006). In addition, HO proteins likely have intrinsic cytoprotective properties (Hori et al, 2002; Kim and Doré, 2005). HO1 catalyzes the cleavage of heme (a pro-oxidant) to form iron (which can then increase ferritin levels), carbon monoxide (a vasodilator and anti-apoptotic agent), and biliverdin, which is rapidly reduced to bilirubin (an antioxidant) (Doré, 2002). Increased synthesis of HO1 leads to the degradation of heme molecules, producing the potent antioxidants biliverdin and bilirubin that are most likely responsible for the neuroprotective actions of HO1, and, therefore, of EC.
In neuronal viability assays, EC reduced neuronal cell death triggered by
In conclusion, we observed that the flavanol EC dose-dependently protected transient ischemia-induced brain injury in both pre- and post-treatment paradigms. The protective mechanism is likely mediated at least in part by the Nrf2/HO1 pathway. These results provide a better understanding of the mechanism by which dietary flavanols can be beneficial and may help promote the design of improved neuroprotective agents for use in stroke.
Footnotes
Acknowledgements
The authors thank Claire Levine for her assistance in the preparation of the paper and all Doré lab members for their insightful contributions.
The authors declare no conflict of interests.