
Abstract
Kinins are proinflammatory and vasoactive peptides that are released during tissue damage and may contribute to neuronal degeneration, inflammation, and edema formation after brain injury by acting on discrete bradykinin receptors, B1R and B2R. We studied the expression of B1R and B2R and the effect of their inhibition on lesion size, blood–brain barrier (BBB) disruption, and inflammatory processes after a focal cryolesion of the right parietal cortex in mice. B1R and B2R gene transcripts were significantly induced in the lesioned hemispheres of wild-type mice (P<0.05). The volume of the cortical lesions and neuronal damage at 24 h after injury in B1R −/− mice were significantly smaller than in wild-type controls (2.5±2.6 versus 11.5±3.9 mm3, P<0.001). Treatment with the B1R antagonist R-715 1 h after lesion induction likewise reduced lesion volume in wild-type mice (2.6±1.4 versus 12.2±6.1 mm3, P<0.001). This was accompanied by a remarkable reduction of BBB disruption and tissue inflammation. In contrast, genetic deletion or pharmacological inhibition of B2R had no significant impact on lesion formation or the development of brain edema. We conclude that B1R inhibition may offer a novel therapeutic strategy after acute brain injuries.
Introduction
Traumatic brain injury (TBI) remains a major cause of disability and death in developed countries (Tagliaferri et al, 2006). In spite of extensive efforts and advances in understanding of the acute pathophysiology of TBI, no specific therapies are available thus far (Beauchamp et al, 2008; Margulies and Hicks, 2009). The initial traumatic insult initiates self-propagating deleterious biochemical and immunologic processes leading to secondary damage characterized by widespread degeneration of neurons and glial cells (Greve and Zink, 2009; Margulies and Hicks, 2009). Key contributing factors to the secondary brain damage are inflammation, metabolic disturbances, and cerebrovascular dysfunction that further propagates injury-induced tissue ischemia and brain edema due to breakdown of the blood–brain barrier (BBB) (Margulies and Hicks, 2009).
The kallikrein–kinin system represents a potential endogenous target to combat injury-induced edema and inflammation (Unterberg et al, 1986; Schilling and Wahl, 1999). All components of the kallikrein–kinin system have been identified in the brain (Camargo et al, 1973; Kariya et al, 1985; Kizuki et al, 1994) and kinins, for example, bradykinin and kallidin constitute the end products of this enzymatic cascade. Kinins are proinflammatory peptides that mediate the classic symptoms of inflammation, vascular, and pain responses to tissue injury by stimulating their two G-protein-coupled receptors, bradykinin receptor B1 (B1R) and B2R (Marceau and Regoli, 2004; Leeb-Lundberg et al, 2005). The tissue expression of B2R is ubiquitous and constitutive (Marceau and Regoli, 2004). This receptor subtype mediates the physiologic effects of bradykinin, whereas the B1R is transiently induced by tissue injury and inflammation. After their activation, both B1R and B2R mediate the classic inflammatory processes after tissue injury such as proinflammatory cytokine release, immune cell influx, and increased vascular permeability (Marceau and Regoli, 2004; Leeb-Lundberg et al, 2005). Previous studies have established a function for the kallikrein–kinin system in the pathophysiology of ischemic injury and TBI (Unterberg et al, 1986; Schilling and Wahl, 1999; Plesnila et al, 2001; Austinat et al, 2009). In these earlier studies, B2R has been characterized as the receptor mediating detrimental effects of kinins after acute brain injury (Gorlach et al, 2001; Plesnila et al, 2001; Thal et al, 2009; Trabold et al, 2010; Zweckberger and Plesnila, 2009). In a recent study (Austinat et al, 2009) we identified the B1R as novel target in experimental stroke. Thus, a dramatic reduction in infarct volume, brain edema, and inflammation was seen in B1R−/− mice subjected to transient middle cerebral artery occlusion (Austinat et al, 2009). These protective actions were also present after pharmacological antagonism of B1R (Austinat et al, 2009). Remarkably, in our previous study no beneficial effect on infarct volumes, brain edema, or inflammation could be observed in B2R-deficient mice or in wild-type mice treated with a selective B2R inhibitor (Austinat et al, 2009). To investigate whether this pattern of bradykinin receptor action could also be observed in another model of acute brain injury, we tested the effects of B1R and B2R deficiency or pharmacological blockade on lesion volume, BBB permeability, and inflammatory processes after a cryogenic cortical trauma, an established brain injury model for profound brain edema and BBB leakage in mice (Sirén et al, 2006; Beauchamp et al, 2008).
Materials and methods
Animals
A total of 142 mice were used in this study. All experiments were approved by and conducted in accordance with the laws and regulations of the regulatory authorities for animal care and use in Lower Franconia. Male 8-week-old B1R−/− and B2R−/− mice backcrossed for more than 10 generations to C57Bl/6 background (Pesquero et al, 2000; Austinat et al, 2009) or C57Bl/6 wild-type mice (Charles River, Sulzfeld, Germany) were used. The weight of mice ranged between 18 and 24 g.
Surgery
We deliberately chose the cryogenic lesion model in mice to address the function of kinin receptors in focal brain trauma. This model is advantageous in comparison to more established models of diffuse TBI (e.g., fluid percussion, cortical impact) in that the lesions are clearly circumscribed and highly reproducible in size and location. Moreover, cryogenic cortical injury has been shown to induce early and profound BBB leakage and inflammation (Sirén et al, 2006), two key readout parameters of the present investigation.
The mice were anesthetized with intraperitoneal injections of ketamine (0.1 mg/g) and xylazine (0.005 mg/g). Surgery was performed on the right parietal cortex after exposing the skull through a scalp incision as described (Sirén et al, 2006). Briefly, a copper cylinder with a tip diameter of 2.5mm was filled with liquid nitrogen (−183 °C) and placed stereotactically on the right parietal cortex (coordinates from bregma: 1.5mm posterior, 1.5mm lateral) for 90 secs. Sham-operated animals went through the same procedure without cooling the copper cylinder. All operations were performed by the same operator (FR) masked to the genotype or treatment group.
The selective B1R inhibitor R-715 (Gobeil et al, 1996) (Ac-Lys-[D-β Nal7, Ile8]-des-Arg9-BK; Biomatik) was administered intravenously 1 h before or after cryolesion at a dosage of 0.5 or 1 mg/kg body weight, respectively. For the selective blockade of B2R, Hoe140 (Wirth et al, 1991) (D-Arg0-Hyp3-Thi5-D-Tic7-Oci8-BK; 0.2 or 0.4 mg/kg body weight; Sigma-Aldrich, Munich, Germany) was injected intravenously 1 h after the induction of the cryogenic lesion. To further address a potential B2R effect in the development of cryogenic lesion injury, we treated B1R−/− mice with the selective B2R inhibitor Hoe140 (0.2 mg/kg body weight intravenously) 1 h after setting of the lesion. Animals were always killed 24 h after surgery for analysis.
Determination of Lesion Size
At 24 h after lesion induction, the brains were quickly removed and cut into six 1-mm-thick coronal sections using a mouse brain slice matrix (Harvard Apparatus, Holliston, MA, USA). The slices were stained with 2% 2,3,5-triphenyltetrazolium chloride (TTC; Sigma-Aldrich) in phosphate-buffered saline (PBS) to visualize the lesions. Planimetric measurements (ImageJ software; National Institutes of Health, Bethesda, MD, USA) of the slices were performed masked to the groups and were used to calculate lesion volumes (Austinat et al, 2009).
Determination of Blood–Brain Barrier Permeability
To determine the permeability of the cerebral vasculature, we intravenously injected 2% Evan's Blue tracer (Sigma-Aldrich) diluted in 0.9% NaCl 2 h after the induction of the cryogenic lesion in R-715- and Hoe140-treated mice or vehicle-treated controls. After 24 h mice were transcardially perfused with 4% paraformaldehyde and brains were quickly removed and cut in six 1-mm-thick coronal sections using a mouse brain slice matrix (Harvard Apparatus). Planimetric measurements (ImageJ software) of the brain parenchyma stained with Evan's Blue were performed to calculate edema volumes.
PCR Studies
RNA was always isolated from a 2-mm-thick coronal brain slice containing the cortical cryogenic lesion (coordinates from bregma: 1.5mm posterior, 1.5mm lateral) at 12, 24, and 48h after injury. Tissue homogenization, RNA isolation, and real-time RT-PCR were performed as described (Austinat et al, 2009). Briefly, total RNAwas prepared with a Miccra D-8 power homogenizer (ART, Mühlheim, Germany) using the TRIzol reagent (Invitrogen, Karlsruhe, Germany) and was quantified spectrophotometrically. Then, 250 μg of total RNA was reversely transcribed with the TaqMan Reverse Transcription Reagents (Applied Biosystems, Darmstadt, Germany) according to the manufacturer's protocol using random hexamers. Relative mRNA levels of cytokines and B1R and B2R were quantified with the fluorescent TaqMan technology. PCR primers and probes specific for murine B1R (assay ID: Mm00432059_s1), B2R (assay ID: Mm00437788_s1), IL-1β (assay ID: Mm004344228_m1), Transforming growth factor (TGF)βb-1 (assay ID: Mm00441724_m1), endothelin-1 (ET-1) (assay ID: Mm 00438656_m1), interferon γ (assay ID: Mm99999071_m1), and tumor necrosis factor (TNF)α (assay ID: Mm00443258_m1) were obtained as TaqMan Gene Expression Arrays (Applied Biosystems). 18s rRNA (TaqMan Predeveloped Assay Reagents for gene expression, part number: 4319413E; Applied Biosystems) was used as an endogenous control to normalize the amount of sample RNA. The PCR was performed with equal amounts of cDNA in the GeneAmp 7700 sequence detection system (Applied Biosystems) using the TaqMan Universal PCR Master Mix (Applied Biosystems). Reactions were incubated at 50°C for 2mins, at 95°C for 10mins followed by 40 cycles of 15 secs at 95°C and 1min at 60°C. Water controls were included to ensure specificity. Each sample was measured in duplicate and data points were examined for integrity by analysis of the amplification plot. The ΔΔCt method was used for relative quantification of gene expression as described (Livak and Schmittgen, 2001; Austinat et al, 2009).
Histology and Immunohistochemistry
Formalin-fixed brains embedded in paraffin from wild-type mice, B1R−/− mice, and B2R−/− mice on day 1 after cryogenic cortical injury were cut into 4-μm-thick sections across the entire lesion. After deparaffinization and rehydration, tissues were stained with hematoxylin and eosin (Sigma-Aldrich). For immunohistochemistry antigen retrieval was achieved by pretreatment with proteinase (P8038; Sigma-Aldrich). Thereafter, endogenous peroxidase activity was blocked with 3% H2O2 in methanol for 15 mins and unspecific binding was prevented by adding 10% bovine serum albumin for 30 mins. For staining of activated macrophages/microglia, we applied a rat anti-mouse F4/80 antibody (BM4008; Acris, Hiddenhausen, Germany) at a dilution of 1:100 in PBS containing 1% BSA overnight at 4°C. Subsequently the slices were incubated with a biotinylated anti-rat IgG (BA-4001; Vector Laboratories, Burlingame, CA, USA) diluted 1:100 in PBS containing 1% BSA for 45 mins at room temperature. The secondary antibody was linked through streptavidin to a biotinylated peroxidase and staining was performed according to the manufacturer's instructions using StreptABComplex/HRP Duet kit (K0492; Dako-Cytomation, Hamburg, Denmark) and 3,3′-diaminobenzidene (Kem-En-Tec Diagnostics, Taastrup, Denmark) and counterstained with aqueous hematoxylin. Negative controls included omission of primary or secondary antibody and gave no signals (not shown); tissue sections from murine spleens served as positive controls (not shown).
Statistical Analysis
Results are presented as mean ± s.d. All data were tested for Gaussian distribution with the D'Agostino and Pearson omnibus normality test and then analyzed by one-way analysis of variance (ANOVA) followed by Bonferroni post hoc test for correction of multiple comparisons. For statistical analysis Prism Graph 4.0 software (GraphPad Software, San Diego, CA, USA) was used. P values < 0.05 were considered statistically significant.
Results
Expression of B1R and B2R in the Lesioned Cortex of Wild-Type Mice
We first analyzed the mRNA expression of B1R and B2R in the ipsilateral cortex of C57Bl/6 mice after cryogenic brain trauma over time (Figure 1). Both receptors were constitutively expressed at low levels in sham-treated animals. B1R and B2R gene transcripts significantly increased after 12 h with a dominant B1R expression (B1R: 11.4 ± 10.0-fold induction, P < 0.05; B2R: 6.4 ± 4.5-fold induction, P < 0.05). Although B1R induction was transient, B2R mRNA expression was still significantly increased at 24 and 48 h after injury (P < 0.05) (Figure 1). Taken together, these data indicate that both kinin receptors are expressed in the murine brain and—just like their ligand bradykinin (Trabold et al, 2010)—undergo induction after brain trauma, suggesting a functional task of the kallikrein–kinin system after focal brain injury.
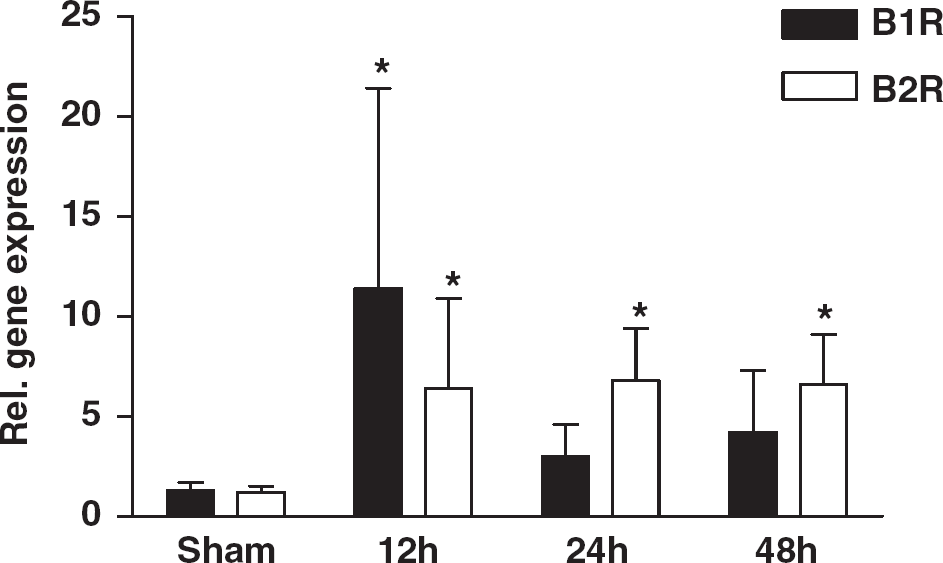
Bradykinin receptor B1 (B1R) and B2R are induced after focal brain damage. Relative gene expression of B1R and B2R in the lesioned cortex of wild-type mice 12, 24, and 48 h after cryogenic cortical injury or sham operation (n = 5 per time point). * P < 0.05, one-way ANOVA, Bonferroni post hoc test compared with sham-operated mice.
Cortical Lesion Volume in B1R−/− Mice, B2R−/− Mice, and Wild-Type Controls
We subjected B1R−/− and B2R−/− mice to cryogenic cortical injury and, after 24h, assessed lesion volumes by staining brain sections with TTC (Figure 2A). B1R−/− mice developed significantly smaller cortical brain lesions compared with wild-type controls (2.5 ± 2.6 versus 11.5 ± 3.9mm3, P < 0.001). In line with these findings, hematoxylin and eosin stainings revealed markedly less neurodegeneration in the cortices of B1R−/− mice than in those of wild-type controls (Figure 2B). In contrast, B2R-deficient mice only showed a tendency toward reduced lesion volumes but the difference did not reach statistical significance (6.5 ± 5.1 versus 11.5 ± 3.9mm3, P > 0.05) (Figure 2A). Previous reports have emphasized the pathophysiological function of B2R in diffuse traumatic brain damage (Gorlach et al, 2001; Plesnila et al, 2001; Hellal et al, 2003; Trabold et al, 2010; Zweckberger and Plesnila, 2009). To further address a potential B2R effect in the development of cryogenic brain injury, we treated B1R−/− mice with the selective B2R inhibitor Hoe140. Application of Hoe140 had no additive benefit on lesion reduction in B1R−/− mice (2.5 ± 2.6 versus 2.8 ± 1.8mm3, P > 0.05) (Figure 2A) hence, making a prominent function of B2R in this model unlikely.
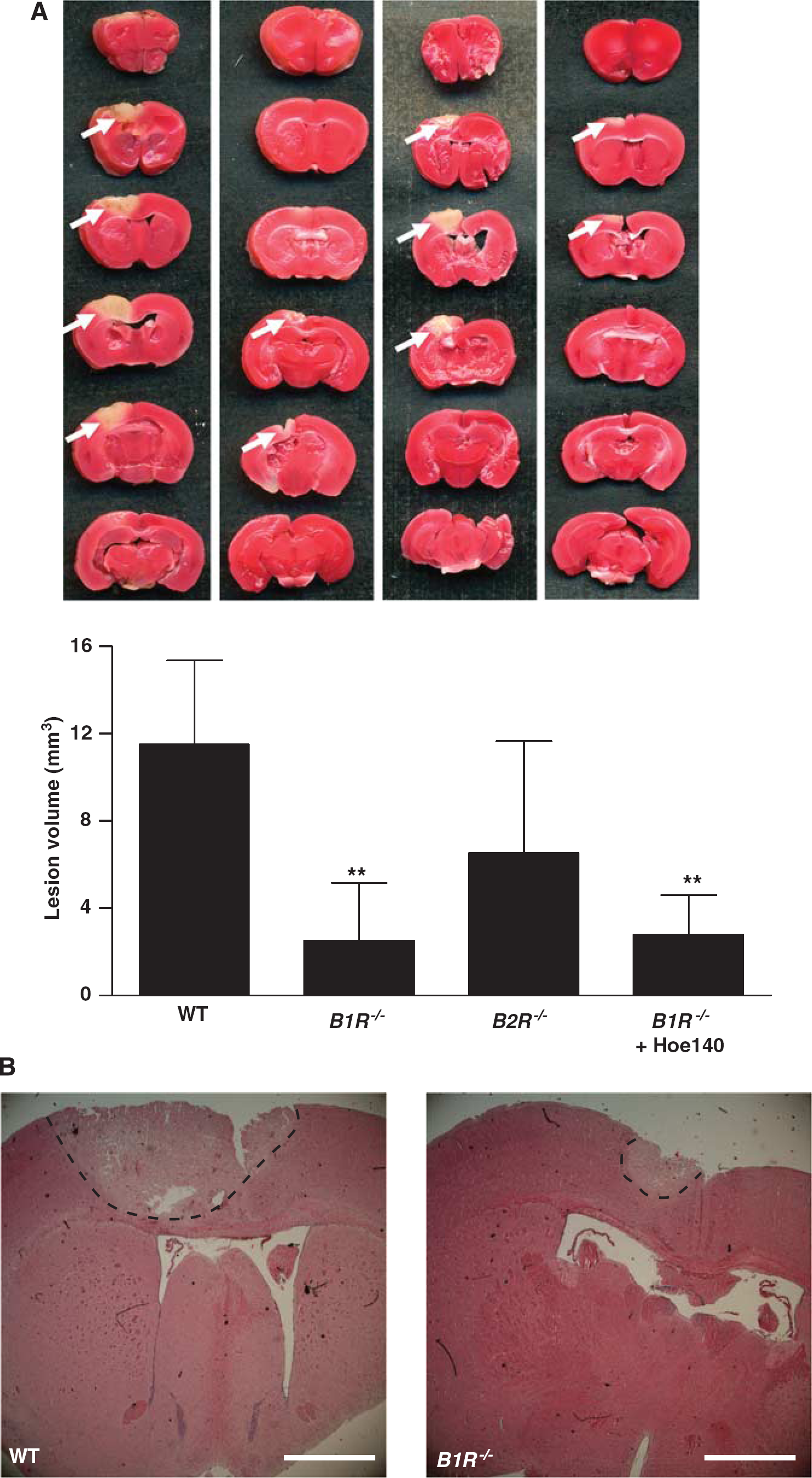
Bradykinin receptor B1 (B1R) deficiency protects from cortical brain damage. (
Effect of a Pharmacological Blockade of B1R and B2R in Wild-Type Mice
As congenital B1R deficiency protected from experimental cryogenic brain lesion, we next tested whether pharmacological targeting of B1R is equally effective. In accordance with the observations made in B1R-deficient mice, the B1R selective blocker R-715 (1 mg/kg body weight) significantly reduced cortical lesion volumes at 24 h when administered as a pretreatment 1 h before setting of the lesion (2.6 ± 1.4 versus 11.7 ± 4.8mm3, P < 0.001) (Figure 3). Importantly, 1mg R-715 per kg body weight was also able to diminish lesion size when applied in a therapeutic approach 1 h after cryogenic brain trauma (3.9 ± 2.4 versus 11.7 ± 4.8mm3, P < 0.001). In contrast, R-715 administered at a decreased dosage of 0.5 mg/kg body weight did not confer neuroprotection after focal brain injury (Figure 3) (P > 0.05). In line with the results obtained in genetically engineered mice (Figure 2), pharmacological B2R inhibition using Hoe140 at a dosage of 0.2 or 0.4 mg/kg had no significant impact on lesion volumes on day 1 (Figure 3) (P > 0.05).
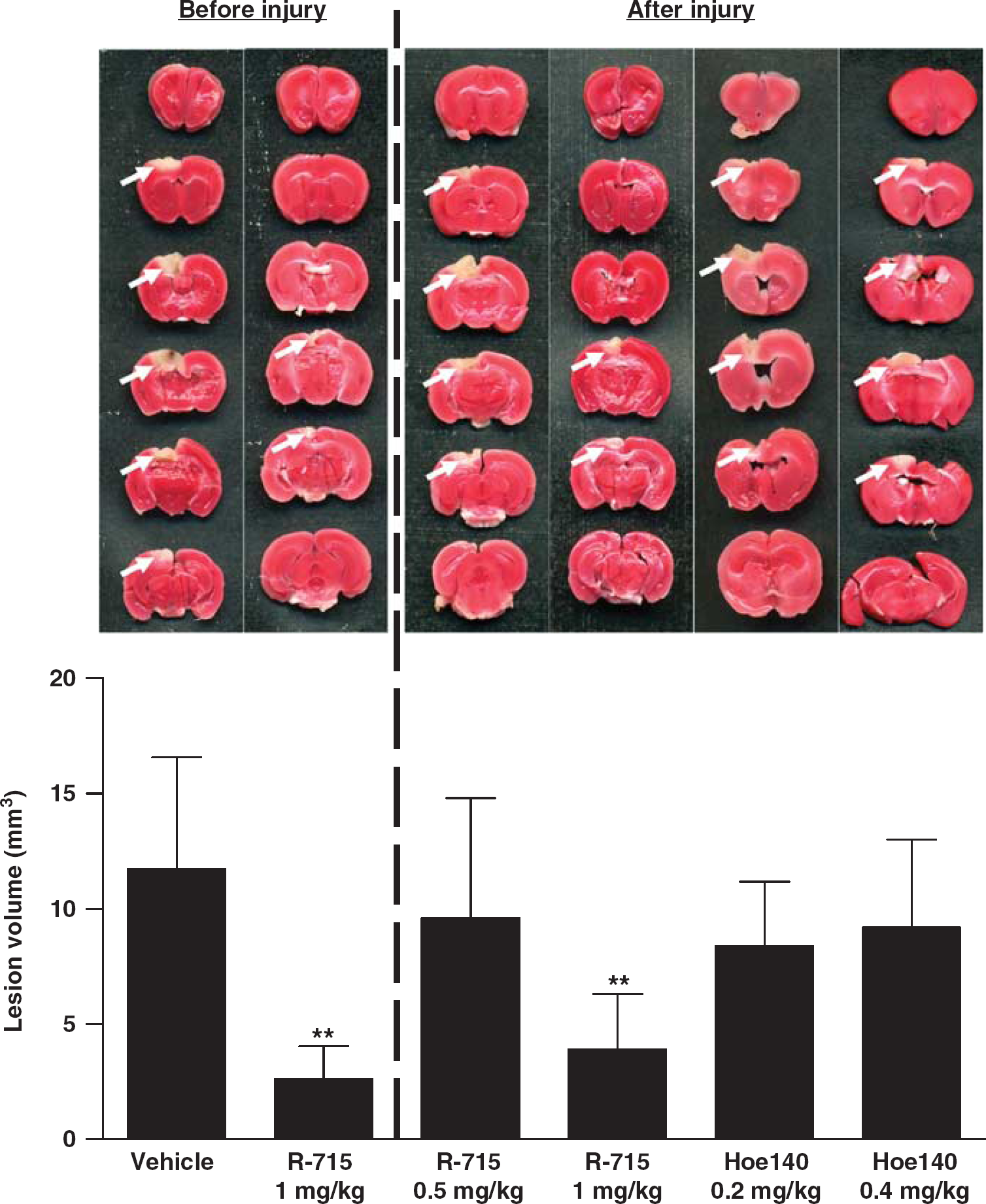
Pharmacological blockade of bradykinin receptor B1 (B1R) but not B2R protects from cortical brain damage. (top, left panel) Representative 2,3,5-triphenyltetrazolium chloride (TTC) stains of six corresponding coronal brain sections 24 h after cryogenic cortical injury from mice treated with the B1R inhibitor R-715 (1 mg/kg) or vehicle 1 h before the operation. (top, right panel) Representative TTC stains of six corresponding coronal brain sections 24 h after cryogenic cortical injury from mice treated with R-715 (1 mg/kg), R-715 (0.5 mg/kg), or the B2R inhibitor Hoe140 (0.2 and 0.4 mg/kg) 1 h after the operation. (bottom) Lesion volumes as assessed by volumetry (n = 8 per group). Note that antagonism of B1R is effective both before or after cortical injury whereas the pharmacological blockade of the B2R is ineffective. **P < 0.001, one-way analysis of variance (ANOVA), Bonferroni post hoc test compared with vehicle-treated mice.
Blockade of B1R Reduces Blood–Brain Barrier Leakage and Inflammation
Edema formation and inflammation critically contribute to secondary tissue damage in the cryogenic brain injury model (Sirén et al, 2006). We therefore studied next the effect of B1R and B2R antagonism on BBB integrity and the expression of vasoactive and proinflammatory mediators in the lesioned brain tissue.
Posttreatment with the selective B1R inhibitor R-715 reduced Evan's blue extravasation, that is, edema formation, in the ipsilateral cortex to approximately 50% as compared with NaCl-treated animals (5.8 ± 4.5 versus 12.5 ± 1.0mm3, P < 0.05), whereas the B2R antagonist Hoe140 had no effect on the lesion-induced increase in brain edema formation (P < 0.05) (Figure 4A). Endothelin-1 is critically involved in the regulation of vascular permeability in the central nervous system and can enhance BBB leakage under pathophysiological conditions (Lo et al, 2005; Kreipke et al, 2010). Accordingly, gene expression of ET-1 was induced on day 1 in lesioned brain tissue of vehicle-treated mice compared with sham-operated controls (3.1 ± 0.4-fold induction, P < 0.001) (Figure 4A). Administration of R-715 1 h after the induction of focal brain injury was able to reduce the amount of ET-1 transcripts to the level observed in the cortices of sham-operated mice (P < 0.001). Again, the B2R antagonist Hoe140 had no effect on ET-1 expression (Figure 4A).
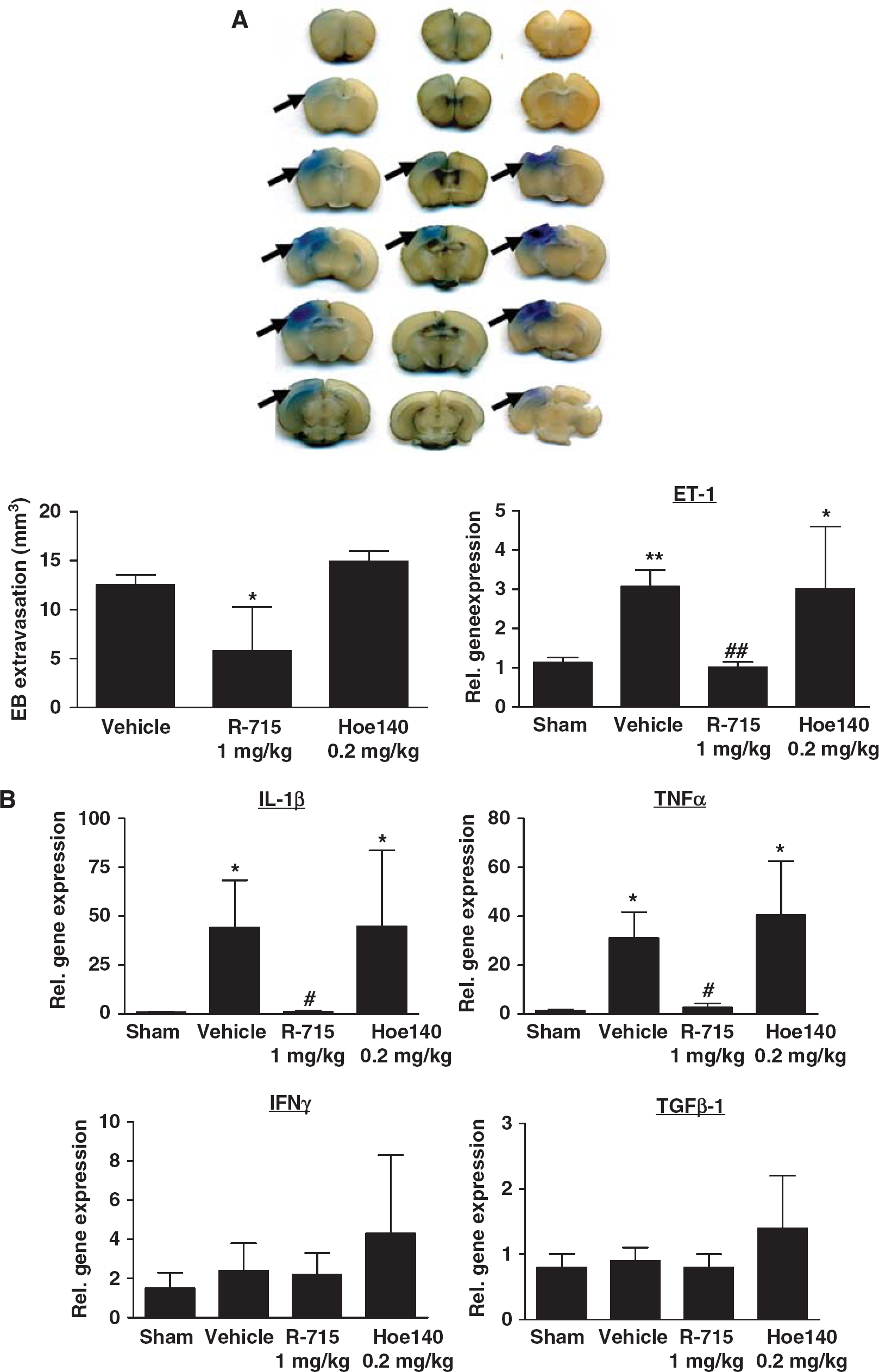
Bradykinin receptor B1 (B1R) blockade but not B2R blockade reduces blood–brain barrier (BBB) leakage and inflammation after focal brain trauma. (
We also investigated the mRNA levels of several prototypic pro- and anti-inflammatory cytokines in the injured brains of R-715- or Hoe140-treated mice after cryoinjury. IL-1β and TNF-α were significantly induced on day 1 in vehicle-treated controls compared with sham-operated mice (IL-1β: 44.3 ± 24.0-fold induction, P < 0.001; TNF-α: 31.1 ± 10.6-fold induction, P < 0.05) (Figure 4B). Posttreatment with R-715 prevented this IL-1β and TNF-α induction (P < 0.05) whereas administration of Hoe140 did not influence proinflammatory cytokine levels. The number of interferon-γ and TGFβ-1 gene transcripts did not differ between R-715- and Hoe140-treated mice or vehicle-treated controls (Figure 4B).
We finally analyzed the extent of the cellular inflammatory response on cryogenic brain damage by immunohistochemistry (Figure 5). Numerous activated F4/80-positive cells had invaded the lesions on day 1 in vehicle- and Hoe140-treated mice. In contrast, the blockade of B1R using R-715 markedly dampened the invasion of activated macrophages/microglia.
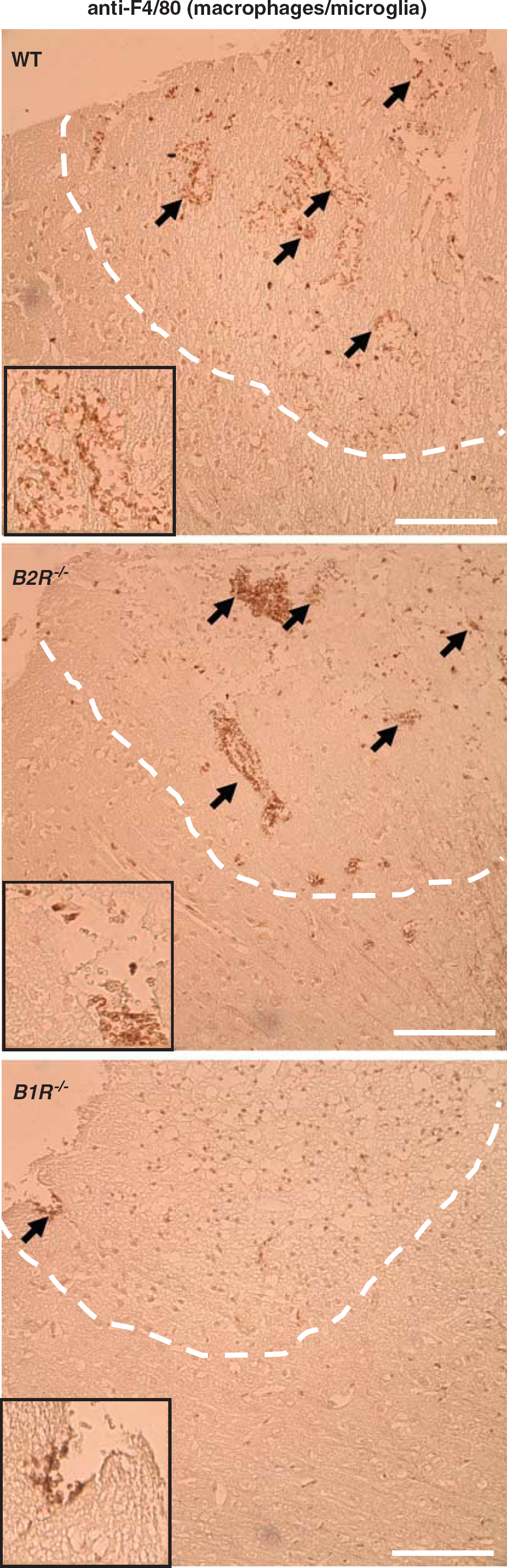
Activated macrophages/microglia in the lesioned cortex (broken white lines) on day 1 after cryogenic brain trauma in wild-type (WT) mice, B1R−/− mice, and B2R−/− mice. Paraffin sections were stained by immunoperoxidase labeling with rat anti-mouse F4/80 antibodies for the detection of activated macrophages/microglia (black arrows). Note that macrophage/microglia invasion was markedly reduced in B1R−/− mice but not in B2R−/− mice compared with WT controls. Bar represents 100 μm (inset: magnification 2.5-fold).
Discussion
Here we show that genetic lack of BR1 receptors or their acute pharmacological antagonism attenuates cortical damage after focal brain injury in mice whereas blockade of B2R has no significant protective effect. Mechanistically, the inhibition of B1R was associated with reduced central nervous system inflammation and BBB leakage after cryogenic cortical injury.
Real-time RT-PCR analysis of the lesioned brain tissue revealed that mRNA expression for both B1R and B2R is induced early after cryogenic injury with a dominating B1R gene expression in the first 12 h after the lesion. Although the induction of B1R was transient, expression of B2R mRNA remained elevated for 48 h. These data are in agreement with previous findings showing upregulation of bradykinin and its receptors after ischemic stroke and head trauma (Unterberg et al, 1986; Schilling and Wahl, 1999; Austinat et al, 2009; Trabold et al, 2010) and suggesting that the kallikrein–kinin system is functionally relevant in the acute phase of focal central nervous system injury. The cells expressing B1R and B2R in the (injured) brain include neurons, microglia, and endothelial cells (Leeb-Lundberg et al, 2005; Trabold et al, 2010).
Previous studies could show that antagonism of the B1R behaves anti-inflammatory and protects from tissue damage under various pathophysiological conditions such as myocardial and renal ischemia (McLean et al, 2000; Lagneux et al, 2002; Souza et al, 2004) or inflammation-induced pain (Pesquero et al, 2000). In addition, we recently established the B1R as novel therapeutic target in ischemic stroke using the transient middle cerebral artery occlusion model in mice that causes widespread hemispheric neurodegeneration and extensive BBB disruption (Austinat et al, 2009). We here further extend these findings by showing that B1R is also critically involved in focal brain trauma after circumscribed cortical cryoinjury. Importantly, the pharmacological B1R blockade was still effective when performed 1 h after lesion induction underlining the therapeutic potential of selective B1R inhibitors some of which are currently under clinical investigation (Rodi et al, 2005).
BBB leakage and subsequent tissue edema are considered important secondary steps in lesion development after cryogenic central nervous system damage (Sirén et al, 2006). Posttreatment with the B1R inhibitor R-715 attenuated the formation of brain edema in this model as indicated by reduced Evan's blue extravasation. Endothelin-1 has been shown to critically influence vascular permeability and tissue perfusion under diverse pathophysiological conditions including brain trauma (Petrov and Rafols, 2001; Iglarz and Clozel, 2007; Kirkby et al, 2008). Antagonism of the ET receptor type A, for example, ameliorated neuronal injury and cerebral hypoperfusion after closed head impact in rats (Kreipke et al, 2010). Moreover, mice overexpressing ET-1 developed more brain edema and larger infarctions on transient middle cerebral artery occlusion (Lo et al, 2005). Interestingly, the induction of ET-1 mRNA in the lesioned hemisphere of wild-type mice was completely suppressed by R-715 after cryogenic cortical injury. In contrast, increased levels of ET-1 were found in animals receiving placebo or B2R antagonist.
Another mechanism by which B1R antagonism probably conveys neuroprotection after focal cortical damage is attenuation of the local inflammatory response. Numerous macrophages/activated microglia cells were present in the brains 24 h after setting of the lesion in wild-type and B2R−/− mice but not in B1R−/− mice. In addition, the expression pattern of different proinflammatory immune mediators was altered: R-715-treated mice produced less IL-1β and TNF-α within the damaged cortex compared with control animals. Both cytokines have been shown to aggravate traumatic brain damage (Morganti-Kossmann et al, 2002).
In contrast to our present results, a number of previous reports have emphasized the function of B2R rather than B1R in the pathophysiology of diffuse TBI (Gorlach et al, 2001; Plesnila et al, 2001; Hellal et al, 2003; Trabold et al, 2010; Zweckberger and Plesnila, 2009). The reasons for this discrepancy are not clear at present. Certainly, the different trauma models used and the pharmacological properties and route of BR-antagonist administration have a function here. One contributing factor may also be the differential pattern of B1R and B2R expression in the various experimental models as a recent study reported no significant change in B2R mRNA expression after controlled cortical impact trauma (Trabold et al, 2010). In accordance with our previous findings in the transient middle cerebral artery occlusion model (Austinat et al, 2009), we observed a strong but transient expression of B1R in the first 12 h after injury whereas the induction of B2R was more sustained lasting up to 48 h.
Taken together, we could show that inhibition of B1R but not B2R protects mice from cortical damage and is associated with reduced BBB leakage and attenuation of the local inflammatory response after cryogenic brain injury. Hence, inhibition of B1R may represent a novel therapeutic approach to combat the detrimental effects of the kallikrein–kinin system after acute brain trauma.
Footnotes
Acknowledgements
We thank Melanie Glaser for excellent technical assistance.
The authors declare no conflict of interest.