
Abstract
P-glycoprotein is an ATP (adenosine triphosphate)-driven drug efflux transporter that is highly expressed at the blood–brain barrier (BBB) and is a major obstacle to the pharmacotherapy of central nervous system diseases, including brain tumors, neuro-AIDS, and epilepsy. Previous studies have shown that P-glycoprotein transport activity in rat brain capillaries is rapidly reduced by the proinflammatory cytokine, tumor necrosis factor-α (TNF-α) acting through protein kinase C (PKC)-dependent signaling. In this study, we used isolated rat brain capillaries to show that the TNF-α-induced reduction of P-glycoprotein activity was prevented by a PKCβI/II inhibitor, LY333531, and mimicked by a PKCβI/II activator, 12-deoxyphorbol-13-phenylacetate-20-acetate (dPPA). Western blotting of brain capillary extracts with phospho-specific antibodies showed that dPPA activated PKCβI, but not PKCβII. Moreover, in intact rats, intracarotid infusion of dPPA potently increased brain accumulation of the P-glycoprotein substrate, [3H]-verapamil without compromising tight junction integrity. Thus, PKCβI activation selectively reduced P-glycoprotein activity both
Keywords
Introduction
P-glycoprotein at the blood–brain barrier (BBB) is a major obstacle to central nervous system (CNS) pharmacotherapy (Miller et al, 2008). This ATP (adenosine triphosphate)-driven efflux transporter limits the ability of a large number of therapeutic drugs to cross the BBB and thus hampers effective treatment of several CNS diseases, including bacterial/viral infection, human immunodeficiency virus encephalitis, epilepsy, multiple sclerosis, neurodegenerative diseases, and brain tumors (Miller et al, 2008). Specific inhibition of P-glycoprotein transport activity (e.g., with cyclosporin A (CSA) or PSC833 (valspodar)) can increase the brain accumulation of clinically important therapeutic compounds in both animal models and humans (Fellner et al, 2002; Hubensack et al, 2008). However, these drugs have consistently failed as chemosensitizers in clinical trials because of significant side effects and organ toxicity (Friedenberg et al, 2006; Hubensack et al, 2008; Pein et al, 2007).
We previously suggested an alternative strategy for improving delivery of P-glycoprotein substrates to the brain: targeting signaling pathways that maintain transporter activity (Hartz et al, 2004, 2006). Studies from this laboratory have shown that exposing isolated rat brain capillaries to tumor necrosis factor (TNF)-α rapidly and reversibly reduces P-glycoprotein transport activity (Hartz et al, 2006). Tumor necrosis factor-α-induced loss of P-glycoprotein activity is signaled through TNF-α receptor 1, endothelin-1 (ET-1), ETB receptor, nitric oxide synthase, and protein kinase C (PKC). The effect of TNF-α or ET-1 is mimicked by treatment with 10 nmol/L PMA (phorbol-12-myristate-13-acetate), and is prevented by treatment with 10 nmol/L BIM-1 (bis-indolylmaleimide), indicating involvement of a classic PKC isoform (α/βI/II/γ) (Hartz et al, 2004, 2006).
Using both
Materials and methods
Reagents
[
Brain Capillary Isolation
All experiments were carried out in compliance with NIH (National Institutes of Health) animal care and use guidelines and approved by the Animal Care and Use Committee of National Institute of Environmental Health Sciences. Brain capillaries were isolated from adult male retired breeder Sprague–Dawley rats as described previously (Hartz et al, 2004). Briefly, rats were killed by CO2 inhalation, decapitated, and their brains were removed immediately to 4°C phosphate-buffered saline (PBS) buffer (in mmol/L: KCl 2.7, KH2PO4 1.5, NaCl 136.9, Na2HPO4 4.3, CaCl2 0.7, MgCl2 0.5, D-glucose 5, and pyruvate 1, pH 7.4). Cortical gray matter was dissected, homogenized, and centrifuged in 15% Ficoll PM 400 for 20 mins (5,800 ×
Transport Assays
Procedures are described in detail in the studies by Bauer et al (2007) and Hartz et al (2006). Briefly, freshly isolated brain capillaries in PBS buffer were transferred into Teflon microscope chambers with glass coverslip bottoms. In the chambers, they were exposed to activators and inhibitors of signaling and to fluorescent substrates (see figure legends for details of individual experiments). In a typical experiment, capillaries were pretreated for 30mins with inhibitors of signaling, followed by exposure to the inhibitors plus TNF-α, ET-1, or dPPA for 1 or 6 h (indicated in the description of each experiment). During the last hour, fluorescent substrates (2 μmol/L NBD-CSA for P-glycoprotein or 2 μmol/L Texas red (sulforhodamine 101) for multidrug resistance associated protein 2 (MRP2) were present in the chamber. We previously showed that 1-h incubation of capillaries with these fluorescent substrates results in steady-state luminal accumulation. Confocal images of capillaries were acquired using a Zeiss 510 meta-laser scanning confocal microscope (Zeiss, Jena, Germany) fitted with a × 40 water-immersion objective (numerical aperture 1.2). Luminal fluorescence intensity was measured as described previously (Hartz et al, 2004; Miller et al, 2000) using NIH Image J software (WS Rasband, Image J, US National Institutes of Health, Bethesda, MD, USA, http://rsb.info.nih.gov/ij/, 1997 to 2008).
Tissue Fractionation
Isolated capillaries obtained from 10 rat brains were divided into 2 to 4 samples. After 30 mins of exposure to dPPA or TNF-α, capillaries were flash frozen in liquid nitrogen and resuspended in 100 μL solution A: PBS (at 4°C) with protease and phosphatase inhibitors (Mini Complete Protease Inhibitor tablet (Roche, Indianapolis, IN, USA) and 2 mmol/L EGTA, 5 mmol/L EDTA, 30 mmol/L NaF, 20 mmol/L pyrophosphate, 10 mmol/L orthovanadate, and 8.6 mg/mL β-glycerophosphate). The suspension was centrifuged at 15,000 ×
Western Blotting
Western blots were performed using the NuPage Bis-Tris electrophoresis system (Invitrogen, Carlsbad, CA, USA). Polyvinylidene fluoride transfer membranes were incubated in Odyssey (Li-Cor, Lincoln, NE, USA) blocking solution for 1 h (room temperature) and incubated overnight at 4°C with primary antibodies. Membranes were washed in PBS with 0.1% Tween and incubated in 50 ng/mL secondary antibody (Odyssey Goat anti-mouse IR Dye 800CW or Goat anti-rabbit IR dye 680CW) for 30 to 60mins. Alternatively, blocking and antibody incubation steps were performed using the Millipore Snap i.d. blotting system according to the manufacturer's specifications (Millipore, Billerica, MA, USA). Western blot infrared fluorescence was detected using the Li-Cor Odyssey infrared imaging system. Integrated band intensities were quantified using the NIH Image J software. For each sample, PKC isoform band intensities were normalized to β-actin and results are expressed as the normalized treatment to control ratio.
Immunohistochemistry
Isolated capillaries adhering to glass coverslips were fixed in 3% paraformaldehyde/0.2% glutaraldehyde for 10 mins, as described previously (Bauer et al, 2007; Hartz et al, 2006). Briefly, fixed capillaries were washed in PBS, permeabilized in 1% Triton X-100 in PBS, and blocked for 1 h with 1% BSA in PBS. Capillaries were incubated in 0.5 μg/mL monoclonal C219 anti-P-glycoprotein antibody (Signet, Dedham, MA, USA) for 1 h at 37°C and washed in 1% BSA, followed by exposure to 2 μg/mL Alexa Fluor 488-conjugated goat anti-mouse IgG (Invitrogen) for 1 h at 37°C. For negative controls, the primary antibody was omitted in the first incubation step. Immunostained capillaries were visualized by confocal microscopy (×60 oil objective; N.A. 1.4), and membrane fluorescence intensity was measured as before (Bauer et al, 2007), using the NIH Image J software.
In Situ Brain Perfusion
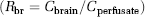
Statistical Analyses
Data are expressed as mean ± s.e. Differences between mean values were deemed statistically significant when
Results
P-Glycoprotein-Mediated Transport in Isolated Rat Brain Capillaries
To determine P-glycoprotein transport activity, we used confocal microscopy and quantitative image analysis to measure steady-state accumulation of NBD-CSA, a fluorescent P-glycoprotein substrate, in the capillary luminal space (Bauer et al, 2007; Hartz et al, 2004, 2006; Miller et al, 2000). As before, luminal accumulation of NBD-CSA was highly concentrative (lumen-to-bath fluorescence ratios exceed 10) and was reduced by NaCN (inhibits energy metabolism) and by the specific P-glycoprotein inhibitor, PSC833 (Figures 1A and 1B). As in previous reports, in this study, we define specific P-glycoprotein transport activity as NaCN- or PSC833-sensitive luminal NBD-CSA accumulation; residual NBD-CSA accumulation represents diffusive entry of NBD-CSA into the luminal space plus nonspecific binding to the capillary tissue (Hartz et al, 2004, 2006; Miller et al, 2000).
LY333531 prevents rapid loss of P-glycoprotein transport activity (steady-state luminal NBD-CSA fluorescence) induced by tumor necrosis factor (TNF)-α and endothelin-1 (ET-1) in rat brain capillaries. (
Rapid Loss of P-Glycoprotein Activity By Tumor Necrosis Factor-α is Signaled Through Protein Kinase C Isoform β
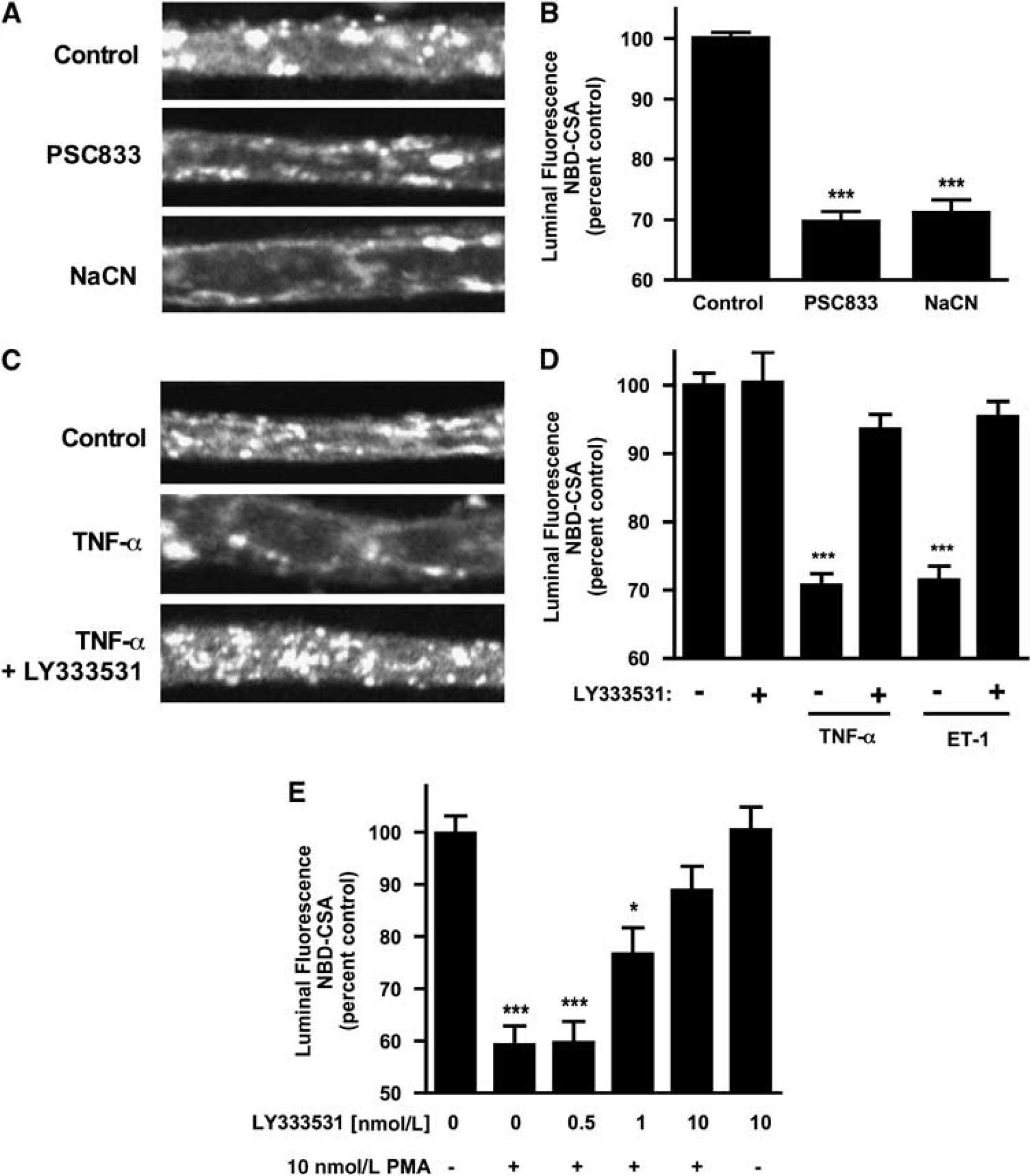
Previous studies have shown reduced P-glycoprotein transport activity in rat brain capillaries exposed to TNF-α or ET-1. (Hartz et al, 2004, 2006). Figures 1C and 1D confirm those findings. For both effectors, the reduction in transport activity was abolished when capillaries were pretreated with LY333531, a specific PKCβI/II inhibitor. LY333531 also abolished the reduction in transport activity caused by exposure to the PKC activator, PMA (Figure 1E). Consistent with PKCβI/II signaling reducing P-glycoprotein transport activity, dPPA, an activator of these PKC isoforms, mimicked the effects of TNF-α, ET-1, and PMA (Figure 2A). dPPA stimulation of transport was antagonized by the isoform-specific inhibitor, LY333531 (Figure 2B). It is noteworthy that the estimated EC50 for LY333531 reversal of dPPA stimulation of transport was 6nmol/L, which agrees well with the half-maximal inhibitory concentration (IC50 = 4 to 6nmol/L) of LY333531 for PKCβI/II (Jirousek et al, 1996). It is also noteworthy that this IC50 is 1 to 2 orders of magnitude lower than that of LY333531 for any other PKC isoform (Jirousek et al, 1996).
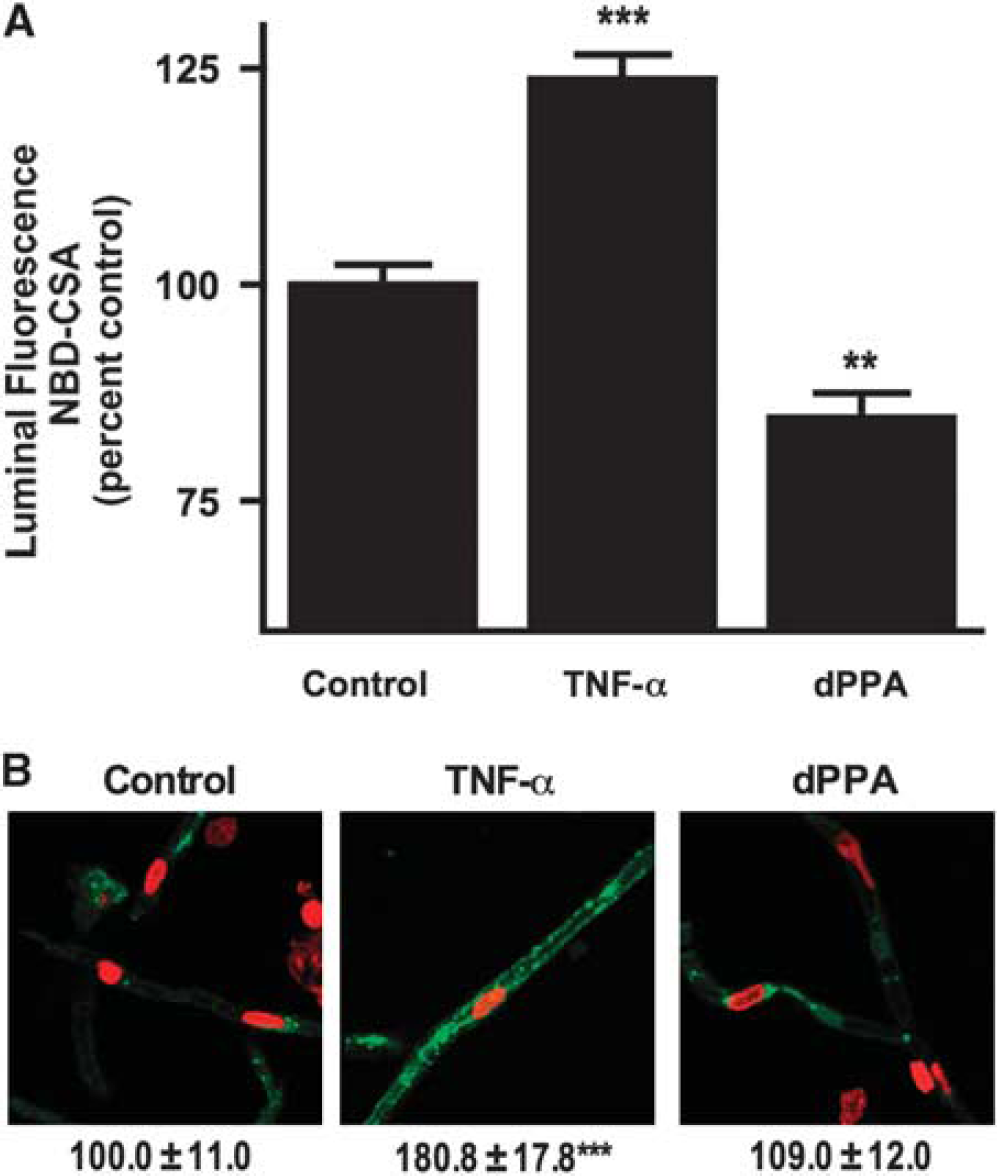
12-Deoxyphorbol-13-phenylacetate-20-acetate (dPPA) reduces P-glycoprotein transport activity.
Although these results are consistent with the loss of specific transport activity, they could also be explained by PKCβI/II activation increasing tight junctional permeability and thus causing leak of pumped NBD-CSA from the luminal space to the bath. We previously showed that tight junction opening did not underlie the reduction in luminal accumulation of NBD-CSA caused by TNF-α or ET-1 (Hartz et al, 2004, 2006). In those experiments, we reasoned that increased tight junctional permeability would affect luminal accumulation of any solute pumped into that space. We showed that neither TNF-α nor ET-1 affected luminal accumulation of the fluorescent organic anion sulforhodamine 101 (Texas red; Hartz et al (2004, 2006)), a substrate for, Mrp2, another ATP-driven efflux pump in rodent brain capillaries, (Bauer et al, 2008a). Figure 2C shows that luminal accumulation of Texas red was not affected by dPPA, but was reduced by the metabolic inhibitor, NaCN and by the addition of 100 mmol/L mannitol to the medium. Exposure of rat brain capillaries to hypertonic mannitol solution increases tight junctional permeability, inducing loss of Texas red and other transported substrates from the luminal space (Hartz et al, 2004, 2006). Thus, dPPA, similar to TNF-α and ET-1, reduced luminal accumulation of NBD-CSA by impairing specific transport rather than by increasing junctional permeability. Taken together, these experiments with isoform-selective PKC effectors identify PKCβI/II as a negative modulator of P-glycoprotein transport activity in rat brain capillaries.
Tumor Necrosis Factor-α Exposure Increases the Amount of Active Protein Kinase CβI/II at the Membrane
Although activation of PKCβI/II was required for the rapid loss of P-glycoprotein activity induced by TNF-α, ET-1, or PMA, the present transport experiments by themselves did not identify which of the two PKCβ isoforms was responsible for the loss of transport, nor the mechanism by which these agents triggered activation of PKC. In general, PKC activation requires translocation of the affected isoform to the plasma membrane, and PDK1-dependent threonine (T500) phosphorylation, followed by autophosphorylation of the isoform. For PKCβI/II, autophosphorylation occurs at serine 661/660 (βI and βII, respectively) and threonine 642/641 (βI and βII, respectively) (Parekh et al, 2000; Steinberg, 2008). Thus, changes in PKC isoform localization and specific residue autophosphorylation could disclose the critical isoform.
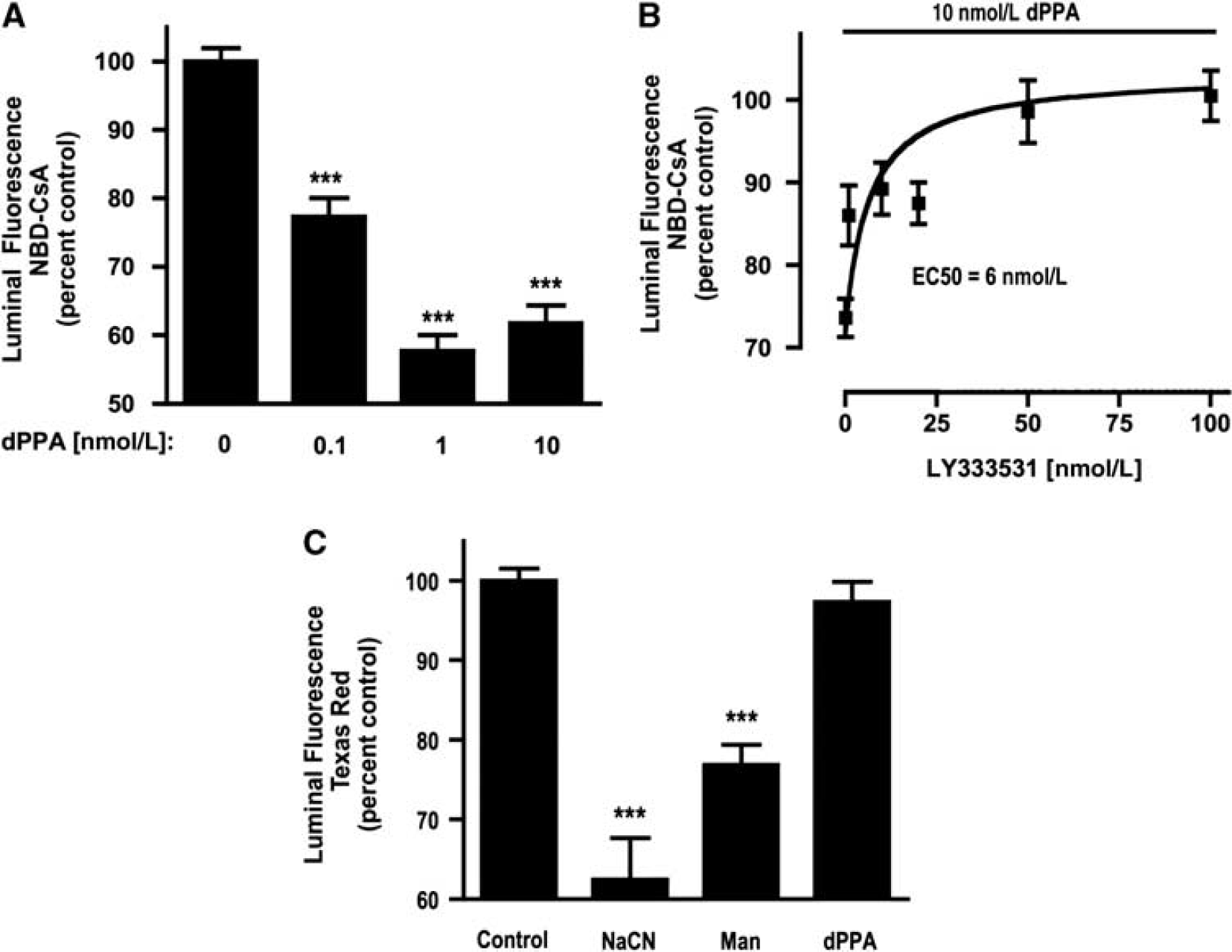
We fractionated brain capillaries to determine the aqueous versus membrane distribution, as well as the autophosphorylation status of PKCβI/II. For these experiments, capillaries were treated with or without (control) 10 ng/mL TNF-α for 30 mins before fractionation. Western blots of aqueous and membrane fractions were probed with antibodies for total PKCα, βI, or βII, as well as for phosphorylated PKCα (S657), βI (T642), or βII (S660). Tumor necrosis factor-α treatment did not change the amounts of total PKCα, βI, or βII in cytosolic or membrane fractions of brain capillaries (for PKCβI or βII, see Figures 3A and 3B; PKCα data not shown). Therefore, TNF-α did not increase protein expression or membrane translocation of PKCα, βI, or βII in brain capillaries. In contrast, TNF-α exposure did increase (by 1.3-fold) the phosphorylation of PKCβI and PKCβII in membrane fractions (Figures 3C and 3D). Both PKCβI and PKCβII phosphorylation levels did not change in aqueous fractions (Figures 3C and 3D), and TNF-α did not increase the phosphorylation of PKCα in aqueous or membrane fractions (not shown). Therefore, rapid inhibition of P-glycoprotein transport activity in response to TNF-α coincided with activation (autophosphorylation) of membrane-localized PKCβI and PKCβII.
Phosphorylation and membrane/aqueous distribution of protein kinase C (PKC)βI (
dPPA Selectively Activates Protein Kinase C Isoform βI in Brain Capillaries
To confirm that dPPA treatment also selectively activated PKCβI/II in brain capillaries, aqueous and membrane fractions of capillary lysates were analyzed by western blotting for total or phosphorylated PKCα, βI, or βII. After 30 mins of exposure to 10nmol/L dPPA, the amount of membrane-associated PKCβI significantly increased 1.5-fold (Figures 4A), indicating that PKCβI translocated to the membrane in response to dPPA treatment. dPPA did not increase the amount of PKCβII (Figure 4B) or PKCα (not shown) at the membrane. dPPA did increase (1.2-fold) the amount of phosphorylated PKCβI at the membrane (Figure 4C). However, phosphorylated PKCβII (Figure 4D) and phosphorylated PKCα (not shown) at the membrane did not increase. Therefore, dPPA treatment specifically activates PKCβI in brain capillaries by translocating only PKCβI (both phosphorylated and unphosphorylated forms) to the membrane.
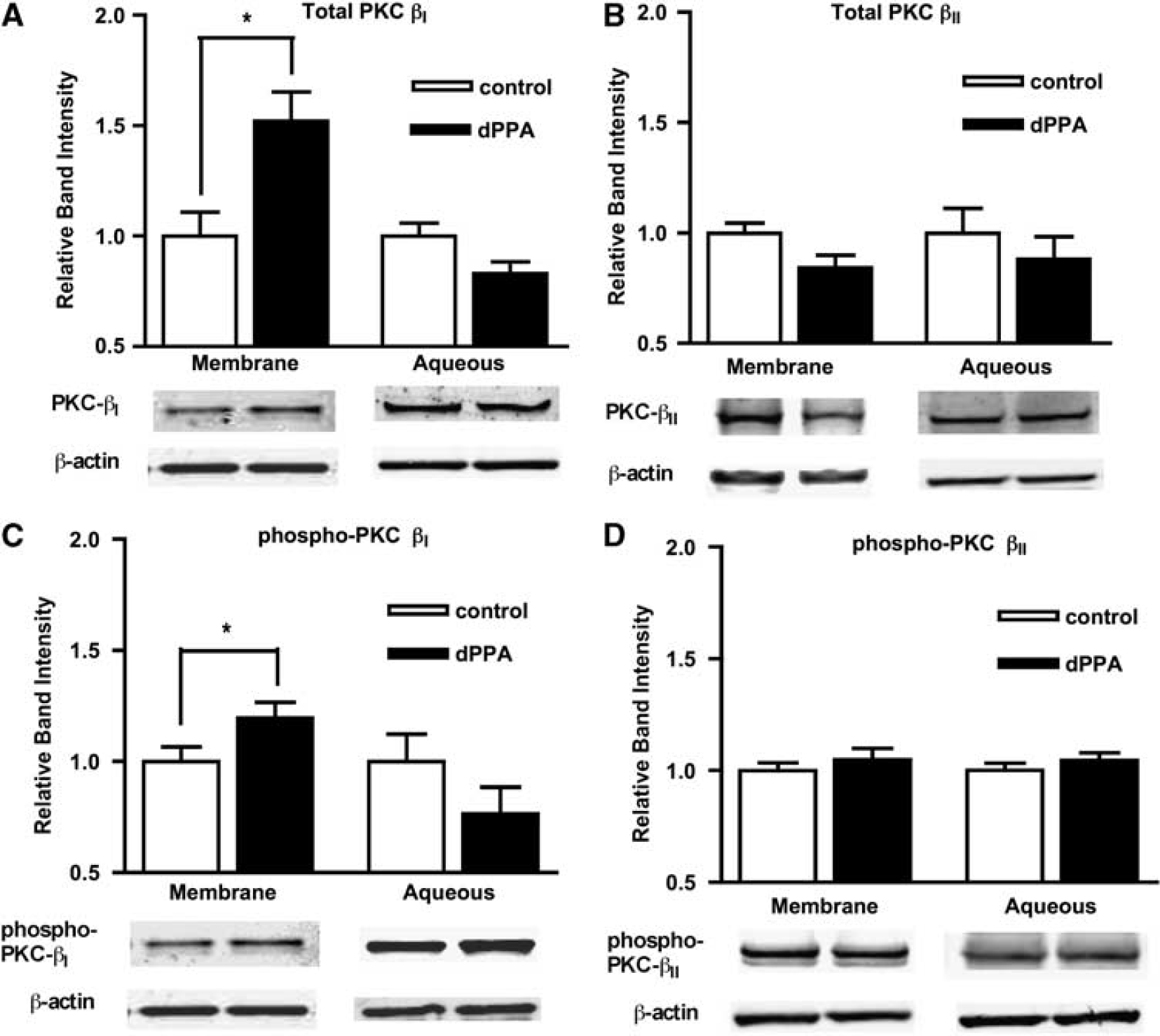
Phosphorylation and membrane/aqueous distribution of protein kinase C (PKC)βI (
dPPA Reduces P-Glycoprotein Activity for at Least 6 h
Tumor necrosis factor-α effects on rat brain capillary P-glycoprotein are time dependent. Tumor necrosis factor-α exposure rapidly reduces P-glycoprotein transport activity (Bauer et al, 2007; Hartz et al, 2006). However, with 3 to 5 h exposure, P-glycoprotein activity recovers to control levels, and with 6 h exposure, P-glycoprotein activity and protein expression substantially exceed control levels (Bauer et al, 2007). As both rapid and long-term effects of TNF-α exposure on P-glycoprotein regulation are inhibited by BIM-1 (Bauer et al, 2007; Hartz et al, 2004, 2006), these multiphasic effects could be mediated entirely by the activation of PKCβI. We exposed capillaries for 6 h to TNF-α or dPPA and then measured P-glycoprotein transport activity and expression. As shown previously (Bauer et al, 2007), 6 h of TNF-α exposure increased P-glycoprotein activity (Figure 5A) and protein expression (Figure 5B). In contrast, 6 h of dPPA exposure caused the same reduction in P-glycoprotein activity observed after 1 h (Figure 5A); immunostaining showed no change in the expression of P-glycoprotein protein after 6 h of dPPA exposure (Figure 5B). Thus, selective activation of PKCβI rapidly reduced P-glycoprotein activity but did not increase transporter expression over the longer term.
P-glycoprotein transport activity (
Activation of Protein Kinase CβI Enhances In Vivo Drug Delivery to the Brain
To test the effects of dPPA exposure on P-glycoprotein transport function
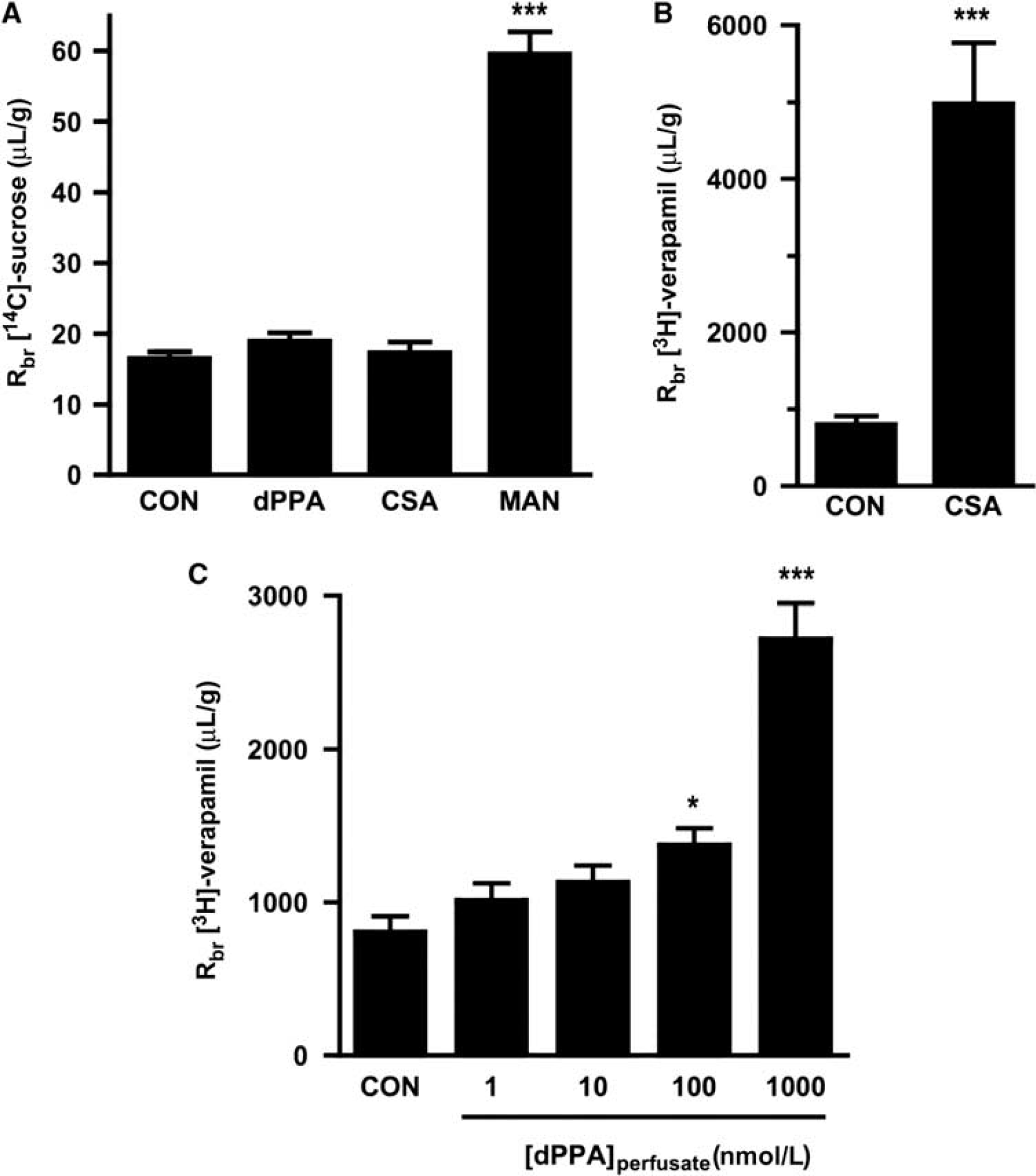
We next measured brain accumulation of [3H]-verapamil, a P-glycoprotein substrate. Similar to previous experiments, 8 μmol/L CSA significantly increased the brain accumulation of [3H]-verapamil 6.2-fold (Figure 6B) (Hawkins et al, 2010). When 1 to 1,000 nmol/L dPPA was added to the perfusate, we observed a concentration-dependent increase in the accumulation of [3H]-verapamil in the brain (Figure 6C). Remarkably, [3H]-verapamil uptake in the brain increased 3.4-fold during the 20 mins perfusion with 1,000 nmol/L dPPA. To confirm that increased brain distribution of [3H]-verapamil was not due to increased accumulation in the endothelium, capillary depletion was performed as described previously (Triguero et al, 1990) on brain samples obtained from a subset of animals in the control and 1,000 nmol/L dPPA groups. We found no significant difference between the radioactivity in brain homogenate and capillary-depleted supernatant in either group, nor was there any significant difference in radioactivity in the capillary pellets between groups. Thus, dPPA substantially enhanced delivery of the drug, [3H]-verapamil, to the brain
Discussion
Limited therapeutic drug delivery to the CNS is a serious problem in the clinic. The ATP-driven drug efflux transporters, e.g., P-glycoprotein, expressed at the BBB substantially underlie our inability to deliver small molecule therapeutics. We previously speculated that TNF-α/ET-1 signaling to BBB P-glycoprotein could be used to rapidly and transiently reduce transporter activity and thus improve brain delivery of drugs that are P-glycoprotein substrates (Hartz et al, 2004, 2006). Such signaling involves TNF-R1 and ETB receptors, nitric oxide synthase, and at least one classic PKC isoform. In this study, we used isolated rat brain capillaries to identify PKCβI as the specific PKC isoform that signals reduced transporter activity and showed that activating that isoform
We found that TNF-α- or ET-1-induced loss of P-glycoprotein transport activity in rat brain capillaries was prevented by a PKCβI/II inhibitor, LY333531, and mimicked by a PKCβI/II selective activator, dPPA. Additional analyses of PKCβI/II translocation and phosphorylation patterns in rat brain capillaries identified PKCβI as the critical isoform activated by both TNF-α and dPPA. Tumor necrosis factor-α increased the phosphorylation of both PKCβI (T642) and PKCβII (S660) in capillary membrane fractions, but had no apparent effect on the aqueous versus membrane distribution of either isoform. dPPA only increased PKCβI membrane translocation and phosphorylation (S657). Neither TNF-α nor dPPA affected phosphorylation or aqueous/membrane distribution of another classic isoform, PKCα. Thus, the rapid loss of P-glycoprotein transport activity induced by TNF-α or dPPA was associated with specific activation of PKCβI, not PKCβII. These findings for PKCβI signaling are novel in two respects: TNF-α has not been shown previously to activate PKCβ;, and PKCβI is not known to regulate P-glycoprotein activity in any tissue. The only reported function of PKCβI signaling in brain capillary endothelium is to increase Na–K–2Cl cotransport activity during hypoxia/aglycemia (Yang et al, 2006), which may contribute to ionic imbalances observed in the brain during ischemia.
It is noteworthy that we previously showed that long-term (6 h) exposure of rat brain capillaries to TNF-α increased both P-glycoprotein transport activity and transporter protein expression (Bauer et al, 2007). The present 6-h experiments confirm the previous finding for TNF-α; but they also show no increases in transport activity or transporter protein expression with 6-h exposure to dPPA. Rather, capillaries exposed to dPPA for 6 h exhibited reduced P-glycoprotein transport activity and control levels of transporter expression, just like capillaries exposed to TNF-α or dPPA for 1 h. As TNF-α signaled through both PKCβI and PKCβII and dPPA only signaled through PKCβI, it appears that the TNF-α-induced increase in P-glycoprotein expression observed after 6 h exposure required activation of PKCβII alone or in concert with PKCβI. At present, we cannot differentiate between these two possibilities. However, it is apparent that the rapid loss of P-glycoprotein transport activity and the delayed increase in transporter expression are signaled through divergent pathways well downstream of TNF-α.
By what sequence of events does PKCβI decrease P-glycoprotein activity in the brain capillary endothelium? There are two aspects to this larger question: how is the signal transmitted to P-glycoprotein, and how is the transporter inactivated? With regard to PKCβI signaling to P-glycoprotein, exhaustive muta-genesis of all consensus PKC phosphorylation sites on P-glycoprotein has shown that transporter phosphorylation by PKC is not required for regulation (Goodfellow et al, 1996). In preliminary experiments, we immunoprecipitated P-glycoprotein from lysates of brain capillaries treated with dPPA and used phospho-serine-, phospho-threonine-, or phosphotyrosine-specific antibodies to detect changes in transporter phosphorylation. No such changes could be detected (RR Rigor, unpublished data). Therefore, rapid loss of P-glycoprotein activity by PKCβI signaling likely involves phosphorylation of an unidentified accessory protein. One such protein is RBCK1, a specific adapter protein and binding partner for PKCβI that confers specificity by linking PKCβI and other regulatory proteins to their targets (Ron and Mochly-Rosen, 1995; Vallentin and Mochly-Rosen, 2007). In preliminary western blot experiments with rat brain capillary lysates, we detected strong expression of RBCK1 (RR Rigor, unpublished data). Identification of RBCK1 binding partners in brain capillaries could help elucidate additional cellular elements responsible for PKCβI-induced loss of P-glycoprotein activity. With regard to transporter inactivation, it is still not clear whether the transporter physically moves to an inactive compartment, e.g., trafficking into an endosome, or whether it is inactivated through protein–protein interactions within the plasma membrane.
The most exciting finding of this study is the observation that pharmacological activation of PKCβI increased by 3.4-fold
It is our contention that a fundamental understanding of the mechanisms that regulate expression and transport activity of P-glycoprotein at the BBB can form the basis for strategies designed to control that transporter in the context of drug delivery. We consider two different ways to target signaling to P-glycoprotein. First, disrupting the signaling pathway through which epileptic seizures increase P-glycoprotein expression has suggested ways to limit that upregulation and thus improve CNS delivery of antiepileptic drugs that are P-glycoprotein substrates, e.g., inhibit cyclooxygenase-2, block EP-1 receptors (Bauer et al, 2008b; Pekcec et al, 2009; Zibell et al, 2009). As this strategy targets signals exclusive to seizure-induced upregulation of transporter expression, its use is limited to that pathway and presumably pathways that intersect upstream of cyclooxygenase-2. Second, activating PKCβI, a signal responsible for establishing the set point for basal transporter activity (in this study) should have wider applicability, especially if the set point can be rapidly and reversibly modulated. It is noteworthy that multiple signaling pathways and transcription factors converge to upregulate P-glycoprotein expression in brain capillaries (Miller et al, 2008). We speculate that targeting transporter activity through PKCβI has the potential to enhance delivery of therapeutic drugs to the brain, even in situations in which transporter expression is upregulated through genomic mechanisms. This exciting possibility remains to be explored.
This
Footnotes
Acknowledgements
The authors wish to thank Destiny Sykes and Jonathan Lucking for excellent technical support.
The authors declare no conflict of interest.