
Abstract
Previous exposure to a nonlethal ischemic insult protects the brain against subsequent harmful ischemia. N-methyl-D-aspartate (NMDA) receptors are a highly studied target of neuroprotection after ischemia. Recently, NMDA receptor subtypes were implicated in neuronal survival and death. We focused on the contribution of NR2A and cyclic-AMP response element (CRE)-binding protein (CREB) signaling to ischemic tolerance using primary cortical neurons. Ischemia in vitro was modeled by oxygen–glucose deprivation (OGD). Ischemic tolerance was induced by applying 45-mins OGD 24 h before 180-mins OGD. Sublethal OGD also induced cross-tolerance against lethal glutamate and hydrogen peroxide. After sublethal OGD, expression of phosphorylated CREB and CRE transcriptional activity were significantly increased. When CRE activity was inhibited by CREB-S133A, a mutant CREB, ischemic tolerance was abolished. Inhibiting NR2A using NVP-AAM077 attenuated preconditioning-induced neuroprotection and correlated with decreased CRE activity levels. Activating NR2A using bicuculline and 4-aminopiridine induced resistance to lethal ischemia accompanied by elevated CRE activity levels, and this effect was abolished by NVP-AAM077. Elevated brain-derived neurotrophic factor (BDNF) transcriptional activities were observed after sublethal OGD and administration of bicuculline and 4-aminopiridine. NR2A-containing NMDA receptors and CREB signaling have important functions in the induction of ischemic tolerance. This may provide potential novel therapeutic strategies to treat ischemic stroke.
Introduction
Stroke is the third leading cause of death worldwide after ischemic heart attack and cancer, and poses a global health threat (Warlow et al, 2003). It is socially important to conquer this disease; however, multiple clinical trials of neuroprotectants to treat stroke patients have failed. In this dead-end situation, ischemic tolerance represents a strategy that may help attenuate ischemic damage. Ischemic tolerance was first demonstrated in 1990 to protect gerbil hippocampal neurons (Kitagawa et al, 1990), and is now a well-established phenomenon in vitro, in vivo, and in clinical settings (Dirnagl et al, 2009; Kirino, 2002). Although it is necessary to better understand ischemic tolerance to overcome this crucial neurologic problem, the molecular mechanisms underlying the phenomenon remain unclear.
The transcription factor cyclic-AMP response element (CRE)-binding protein (CREB) mediates diverse responses in the nervous system, including learning, memory, neuronal plasticity, and cell survival (Barco et al, 2002; Kandel, 2001; Lonze and Ginty, 2002). Phosphorylation of serine-133 in CREB allows it to contact its co-activator, CREB-binding protein/p300, and is necessary for activation. We previously reported that CRE-mediated gene expression increased in the penumbra using CRE-LacZ transgenic mice. The CREB activation was a critical event in neuroprotection against ischemic injury and in ischemic tolerance (Meller et al, 2005; Sugiura et al, 2004).
Meanwhile, the calcium–glutamate hypothesis suggests that excessive release of glutamate during and after ischemic injury leads to neuronal toxicity and death. The N-methyl-D-aspartate (NMDA) type glutamate receptors are associated with ischemic neuronal injury and ischemic tolerance (Grabb and Choi, 1999). On theoretical grounds, central nervous system disorders, including ischemic stroke, caused by glutamate-induced excitotoxicity might be treatable by blocking NMDA receptors. However, clinical application of NMDA receptor antagonists has not succeeded (Ikonomidou and Turski, 2002). Among a number of possible reasons, this is partly because there are multiple NMDA receptor subtypes, which may have different functions. The NMDA receptors are heteromeric complexes incorporating different subunits of the NR1, NR2, and NR3 subtypes. These tetramers most often incorporate two NR1 and two NR2 subunits, of which there are four different isoforms (NR2A to NR2D) (Paoletti and Neyton, 2007). Recent studies suggest that NR2A- and NR2B-containing NMDA receptors may have different functions in epileptogenesis and ischemic stroke (Chen et al, 2008; Chen et al, 2007; Liu et al, 2007). Synaptic NMDA receptors mainly dominated by NR2A were also shown to promote neuronal survival via CREB signaling (Hardingham et al, 2002).
In light of recent reports, we hypothesized that NR2A-containing NMDA receptors and CREB signaling might have a critical function in the induction of ischemic tolerance. In this study, we established ischemic tolerance in cultured cortical neurons and found that activation of NR2A induced ischemic tolerance via CREB signaling.
Materials and methods
The experimental procedures were approved by the Institutional Animal Care and Use Committee of the Osaka University Graduate School of Medicine.
Primary Neuronal Cell Culture
Cortical neuronal cultures were prepared from 16-day-old Wistar rat embryos (Charles River, Yokohama, Japan) using methods described earlier (Mabuchi et al, 2001). Briefly, cortices were dissected and dissociated using papain dissociation system (Worthington Biochemical Corporation, Lakewood, NJ, USA). Cells were plated on dishes coated with polyethylenimine in high-glucose DMEM (Sigma, St Louis, MO, USA) containing 5% fetal bovine serum (Invitrogen, Carlsbad, CA, USA) and 1% Antibiotic-Antimycotic (Invitrogen) at a density of 7 × 105 per cells/mL. At 24 h after seeding, the medium was changed to Neurobasal medium (Invitrogen) supplemented with B-27 (Invitrogen) and 1% Antibiotic-Antimycotic. Cells were cultured at 37°C in a humidified chamber of 95% air and 5% CO2. Cultures were used for experiments 10 to 12 days after seeding.
Oxygen–Glucose Deprivation
For oxygen–glucose deprivation (OGD)/reoxygenation, cells were washed with phosphate-buffered saline and incubated with glucose-free Earle's balanced salts solution medium (Biological Industries Israel Beit Haemek Ltd, Beit Haemek, Israel) in an anaerobic chamber containing 5% CO2 and 95% N2 (< 1% O2). The OGD was terminated by bringing the cultures back to the original medium and placing them in a normoxic chamber. Sublethal ischemia was applied by OGD for 45 mins, and lethal ischemia was applied for 180 mins. For cross-tolerance experiments, cells were treated with 100 μmol/L glutamate or 50 μmol/L hydrogen peroxide for 15 mins. After treatment, neurons were incubated in the original medium.
To investigate the involvement of NR2A in ischemic tolerance, cultures were pretreated for 2 h and treated between sublethal and lethal OGD with one of the following chemicals: NMDA receptor antagonist D(−)-2-Amino-5-phosphopentanoic acid (APV) (100 μmol/L), non-NMDA receptor antagonist CNQX (10 μmol/L), L-type Ca2+ channel blocker nifedipine (5 μmol/L), NR2A-specific antagonist NVP-AAM077 (200 nmol/L), and NR2B-specific antagonist Ro 25-6981 (100 nmol/L). Neurons were not coincubated with these drugs during sublethal OGD, during and after lethal OGD. The potency and specificity of NVP-AAM077 were previously reported that it indicated a 100-fold preference for NR2A-containing receptors over NR2B-containing receptors (Auberson et al, 2002; Mallon et al, 2005). To stimulate neuronal synapses, neurons were pretreated for 24 h before lethal OGD with (−)-bicuculline methiodide (30 μmol/L), a GABAA antagonist, and 4-aminopyridine (4-AP, 2500 μmol/L), a weak potassium channel blocker. NVP-AAM077 was a kind gift from Dr Yves Auberson, Novartis. Ro25-6981 and (−)-bicuculline methiodide were purchased from Tocris (Ellisville, MO, USA). Other agents were purchased from Sigma.
Cell Viability Assays
Neuronal injury was measured 24 h after treatment by lactate dehydrogenase (LDH) using the Cytotoxicity Detection Kit (Roche Applied Science, Mannheim, Germany). In sister culture, 100% cell death was induced with 2 mmol/L NMDA. The relative assessments of neuronal injury were normalized by comparison with 100% cell death.
Adenovirus Transfection and Measurement of Cyclic-AMP Response Element Transcriptional Activity
To investigate an association of CREB phosphorylation with ischemic tolerance, a dominant negative CREB mutant in which serine-133 was mutated to alanine (CREB-S133A, M-CREB) was used. Neurons were transfected with recombinant adenovirus vectors expressing CREB-S133A (adeno-CREB-S133A) 72h before experiments.
The CRE transcriptional activity was quantified by luciferase assay. As plasmid-based reporters did not provide an adequate level of activity in the neurons, we prepared an adenovirus-mediated reporter system. Cortical neurons were transfected with purified adenovirus, adeno-CRE-Luc (firefly) and adeno-TK-Luc (renilla). As an internal control, an adenovirus driving renilla luciferase was used (Promega, Madison, WI, USA). Luciferase activities were measured using the Dual-Luciferase Reporter Assay System (Promega). For luciferase assay, cell lysates were collected with 1 × passive lysis buffer (Promega). The relative firefly luciferase-based CRE-reporter activities were standardized to the corresponding Renilla luciferase in all cases.
Construction of a Mouse Brain-Derived Neurotrophic Factor Exon IV Promoter-Luciferase Reporter
Mouse genomic DNA was used as a template for polymerase chain reaction to generate an ∼400 bp brain-derived neurotrophic factor (BDNF) exon IV fragment with the following primers: 5′-GAGCTCGCCCTCCAGAACCTAGT CA-3′ and 5;-AAGCTTAGAGCAGTCCTCTCCTCGG-3′. The fragment was cloned into the Sac I/Hind III sites of the pGL4 luciferase reporter vector (Promega). A BDNF exon IV promoter-luciferase mutant lacking a CRE motif was generated by exchanging nucleotides using Quick-change Mutagenesis Kit (Stratagene, Cedar Creek, TX, USA) as follows: consensus CRE, T
Western Blot Analysis and Immunohistochemistry
Whole-cell protein extracts of cortical neurons were harvested and prepared for Western blot analysis as described earlier (Mabuchi et al, 2001). Proteins were extracted in lysis buffer composed of 20 mmol/L Tris-HCl, 150 mmol/L NaCl, 2 mmol/L EDTA, 1% Nonidet P-40, protease inhibitor cocktail (Complete Mini, Roche Applied Science) and phosphatase inhibitor cocktail (PhosSTOP, Roche Applied Science). After mixing and boiling for 5 mins with 2x sample buffer, an equal amount of protein for each sample was separated by 7.5% SDS-PAGE, and transferred onto polyvinylidine difluoride membrane (Millipore, Billerica, MA, USA). Blots were blocked in phosphate-buffered saline and 0.1% Tween-20 containing 5% skim milk for 1 h and probed with anti-phospho-CREB (1:1000; Upstate, Temecula, CA, USA), anti-CREB (1:1000; Cell Signaling Technology, Beverly, MA, USA), anti-NMDAR2A (1:1000; Calbiochem, La Jolla, CA, USA), anti-NMDAR2B (1:1000; Chemicon, Temecula, CA, USA) anti-GluR2 (1:1000; BD Biosciences Pharmingen, San Diego, CA, USA), and anti-β-actin (1:100000; Sigma) in Can Get Signal Solution 1 (Toyobo Co. Ltd, Osaka, Japan) at 4°C overnight. The membranes were washed three times in phosphate-buffered saline and 0.1% Tween 20 and incubated for 1 h with anti-rabbit IgG conjugated to HRP (1:5000; Amersham Pharmacia Biotech, Uppsala, Sweden) for anti-phospho-CREB, anti-CREB, anti-NMDAR2A, and anti-NMDAR2B, or with anti-mouse IgG conjugated to HRP (1:2000; Amersham Pharmacia Biotech) for anti-GluR2 and anti-β-actin in Can Get Signal Solution 2 (Toyobo). Blots were developed by enhanced chemiluminescence (GE Healthcare UK limited, Buckinghamshire, UK) and exposed to x-ray film. Digital images were produced by densitometric scans of autoradiographs and quantified by Image J 1.40 software (National Institutes of Health, Bethesda, MD, USA).
For immunohistochemical studies, neurons were fixed in 4% paraformaldehyde for 30 mins, washed and permeabilized in 0.01% Triton X-100 for 30 mins. After blocking with 10% donkey serum, cells were incubated with anti-microtubule-associated protein 2 (MAP2) (1:500; Sigma) at 4°C overnight. Neurons were washed, probed with Alexa Fluor 488 conjugated donkey anti-mouse secondary anti-body (1:300; Invitrogen) at room temperature for 30 mins, and viewed using a confocal microscope.
Statistical Analysis
Data in the text and figures are described as mean ± s.d. Statistical analysis was performed by one-factor analysis of variance followed by Scheffe's post hoc test. Statistical significance was defined as P < 0.05.
Results
Ischemic Tolerance and Cross-Tolerance In Vitro
To elucidate the mechanisms of induction of ischemic tolerance, we established an ischemic tolerance model using in vitro OGD. With regard to ischemic preconditioning (+) group, the cultures were subjected to 45-mins OGD before 180-mins OGD. The medium of ischemic preconditioning (−) group was changed as sham wash instead of 45-mins OGD (Figure 1A). When cells were subjected to 180-mins OGD, we observed ∼70% cell death. When the cultures were preconditioned with a 45-mins OGD before the 180-mins OGD, cell death was reduced (Figure 1B). The most effective interval between sublethal and lethal OGD to obtain ischemic tolerance was 24 h (Figure 1C), whereas, at intervals of 1 and 4 h, the protective effect was not observed. Importantly, the preconditioning ischemia alone did not induce cell death (data not shown). Interestingly, we observed cross-tolerance in these cultures: sublethal OGD reduced neuronal injury by lethal insults such as 15 mins treatment with 100 μmol/L glutamate or 50 μmol/L hydrogen peroxide (Figures 1G–1H).
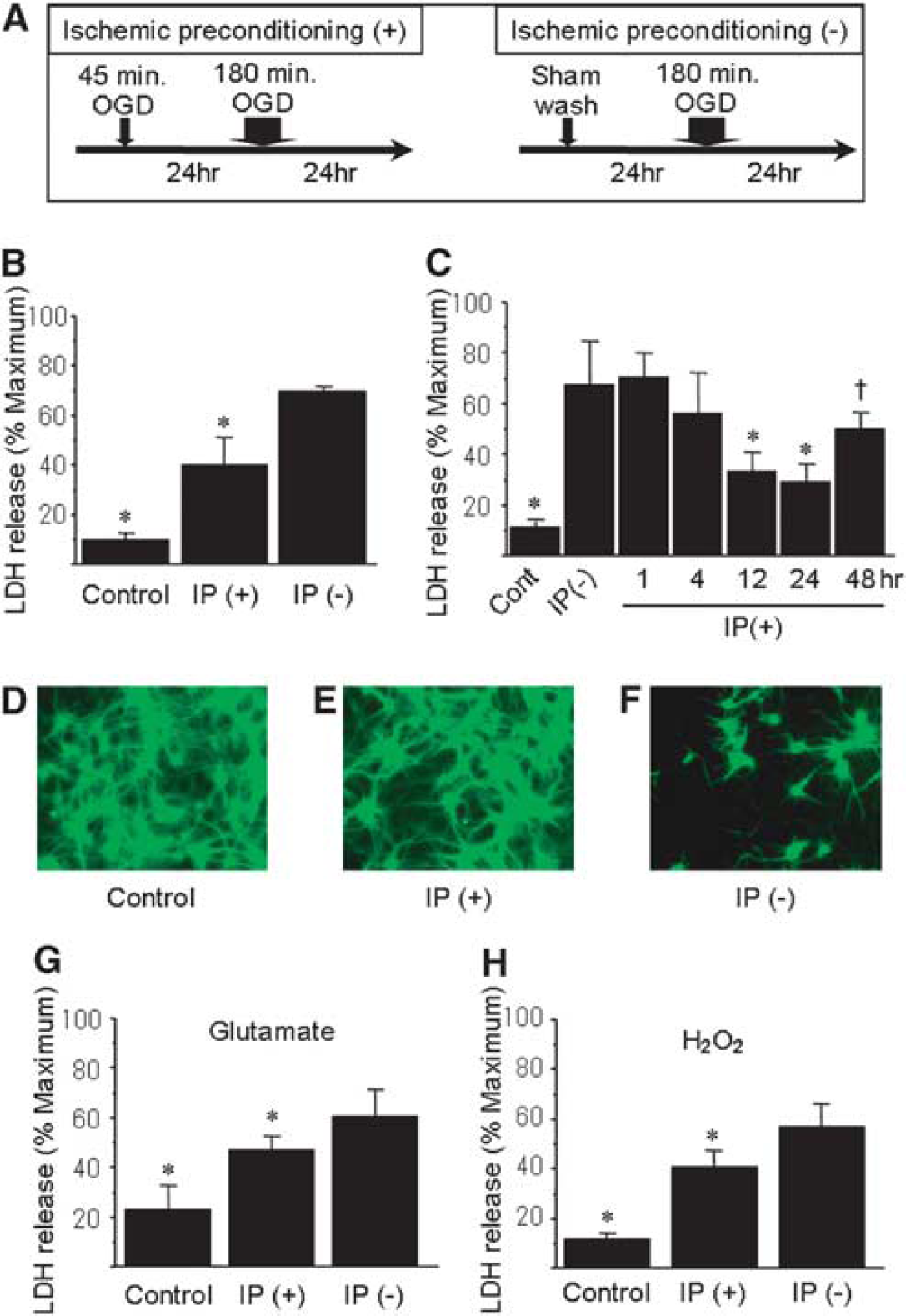
Model of ischemic tolerance and cross-tolerance in primary neuronal cell culture. (
Considering these results, we found that preconditioning OGD could induce both ischemic and cross-tolerance.
Phosphorylation of Cyclic-AMP Response Element-Binding Protein and Cyclic-AMP Response Element Activation are Necessary for Ischemic Tolerance
It has been shown earlier that CREB was activated after ischemic preconditioning (Grabb and Choi, 1999); accordingly, we investigated the effects of preconditioning ischemia on phosphorylation of CREB and CRE transcriptional activity. The CREB was expressed in control cells, and total CREB expression levels did not change after 45-mins OGD. In contrast, phosphorylated CREB levels significantly increased 1 to 6 h after 45-mins OGD (Figure 2A). In the same time course, transcriptional activity of CRE was markedly enhanced 1 h after sublethal OGD, and maintained at 24 h (Figure 2B).
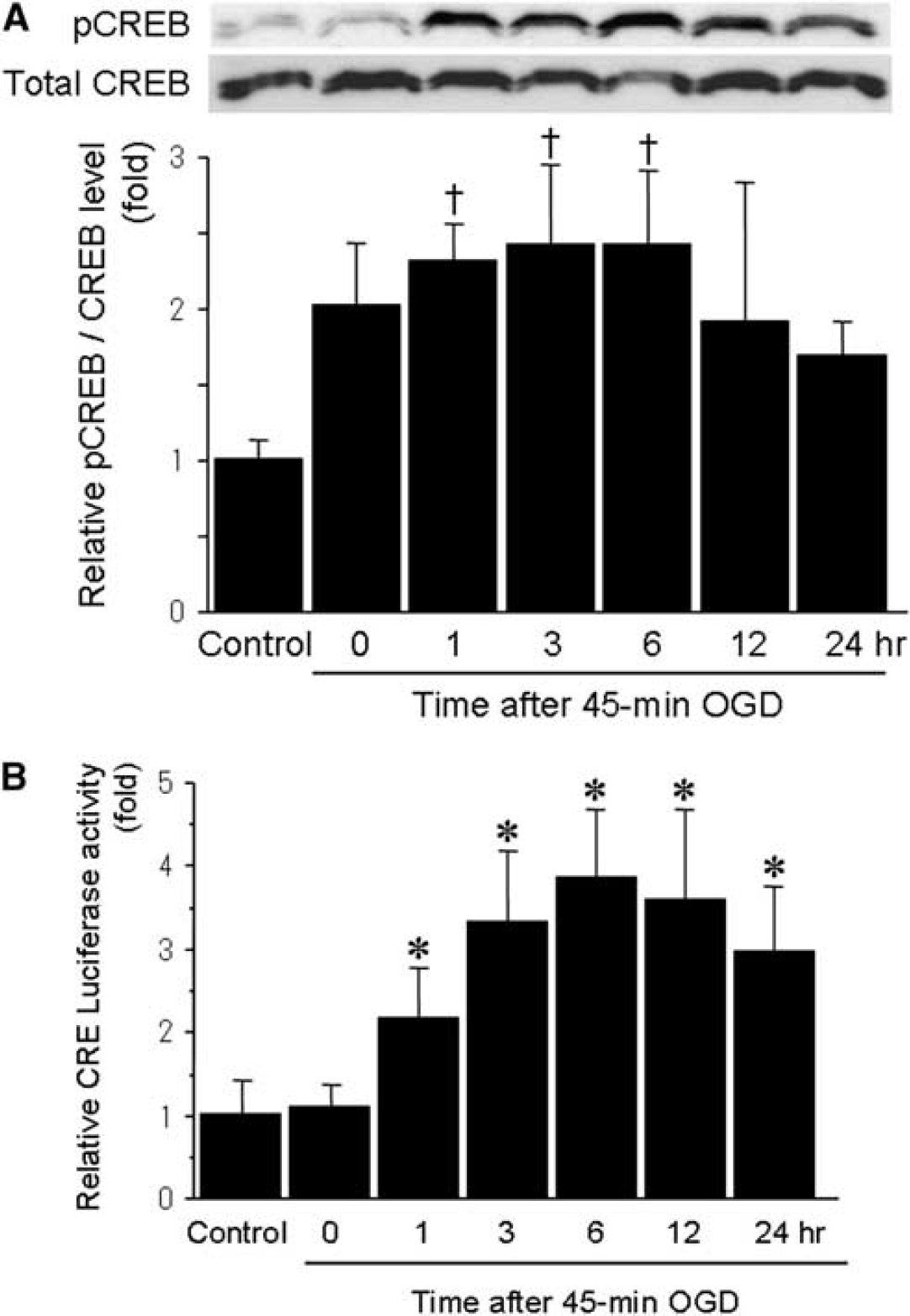
Temporal profiles of expression of phosphorylated CREB and CRE transcriptional activity after sublethal ischemia. (
To further investigate the importance of the CREB cascade, we examined whether a CREB-S133A mutant would affect CRE activity under these conditions. When cells were transfected with adeno-CREB-S133A, CRE transcriptional activity was lower compared with the EGFP-transfected control group after sublethal OGD (Figure 3A). Under these conditions, the adeno-CREB-S133A abolished ischemic tolerance (Figure 3B). In contrast, the EGFP controls showed ischemic tolerance.
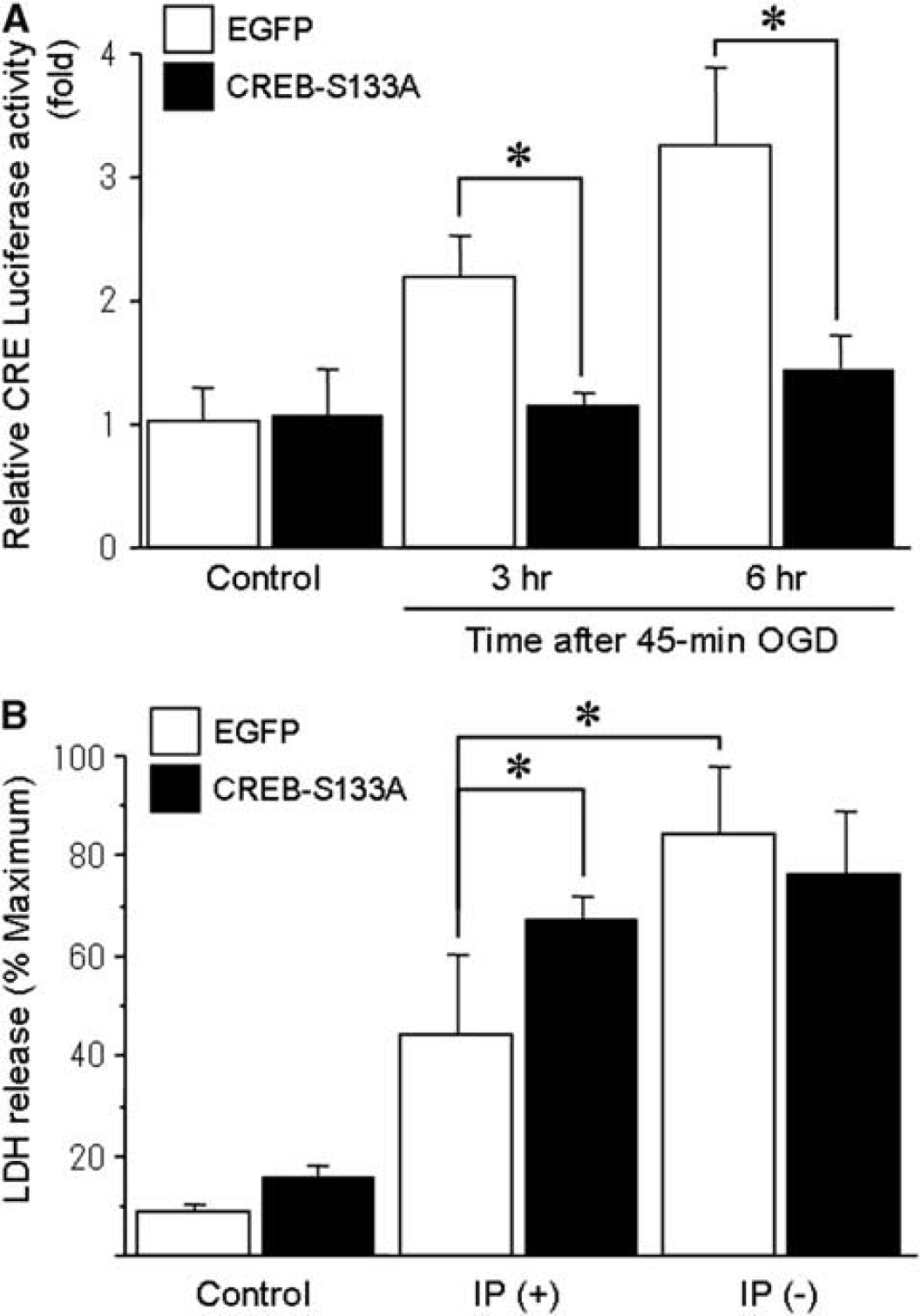
Effect of CREB-S133A on CRE reporter activity and ischemic tolerance. (
Consistent with earlier reports (Chen et al, 2008; Meller et al, 2005), these findings suggest that phosphorylation of CREB and the subsequent enhancement of CRE activity are necessary for induction of ischemic tolerance.
NR2A Activation Protects Neurons Against Lethal Ischemia via Cyclic-AMP Response Element-Binding Protein Signaling
Among the routes of Ca2+ entry, NMDA receptors are of particular interest because of their ability to gate high levels of Ca2+ influx, and they have been extensively studied for their critical function in ischemia. Recently, several studies have suggested that NMDA receptor subtypes (NR2A/synaptic or NR2B/extrasynaptic) are key regulators of neuronal viability (Hardingham et al, 2002). To determine whether different types of glutamate receptor are associated with ischemic tolerance and CREB signaling, we administered several types of glutamate receptor antagonist (Figure 4A). First, we examined the effect of sublethal OGD on the expression of NMDA and AMPA receptors with Western blot analysis. The expression levels of NR2A, NR2B, and GluR2 did not change after sublethal OGD (Figure 4B). Under such a condition, inhibiting NR2A-containing NMDA receptors using NVP-AAM077 attenuated preconditioning-induced neuroprotection (Figure 4C). In contrast, other types of glutamate receptor antagonist such as APV, CNQX, and Ro25-6981, or an L-type calcium channel blocker, nifedipine, did not have much effect on ischemic tolerance. Treatment with these drugs alone exerted little effect on the basal LDH levels. Among the neuronal signaling pathways, NR2A-containing NMDA receptors have been shown to activate the CREB cascade (Hardingham et al, 2002). To further elucidate the pathway downstream of NR2A receptors, we investigated the effect of inhibiting NR2A receptors on phosophorylation of CREB. When cells were treated with NVP-AAM077, phosphorylated CREB levels after sublethal OGD were significantly attenuated compared with those treated with vehicle (Figure 4D). Furthermore, we examined the association between antagonizing NR2A receptors and CRE transcriptional activity. Compared with vehicle treatment, inhibition of NR2A receptors by NVP-AAM077 partially but significantly decreased CRE activity 3 and 6 h after 45-mins OGD (Figure 4E).
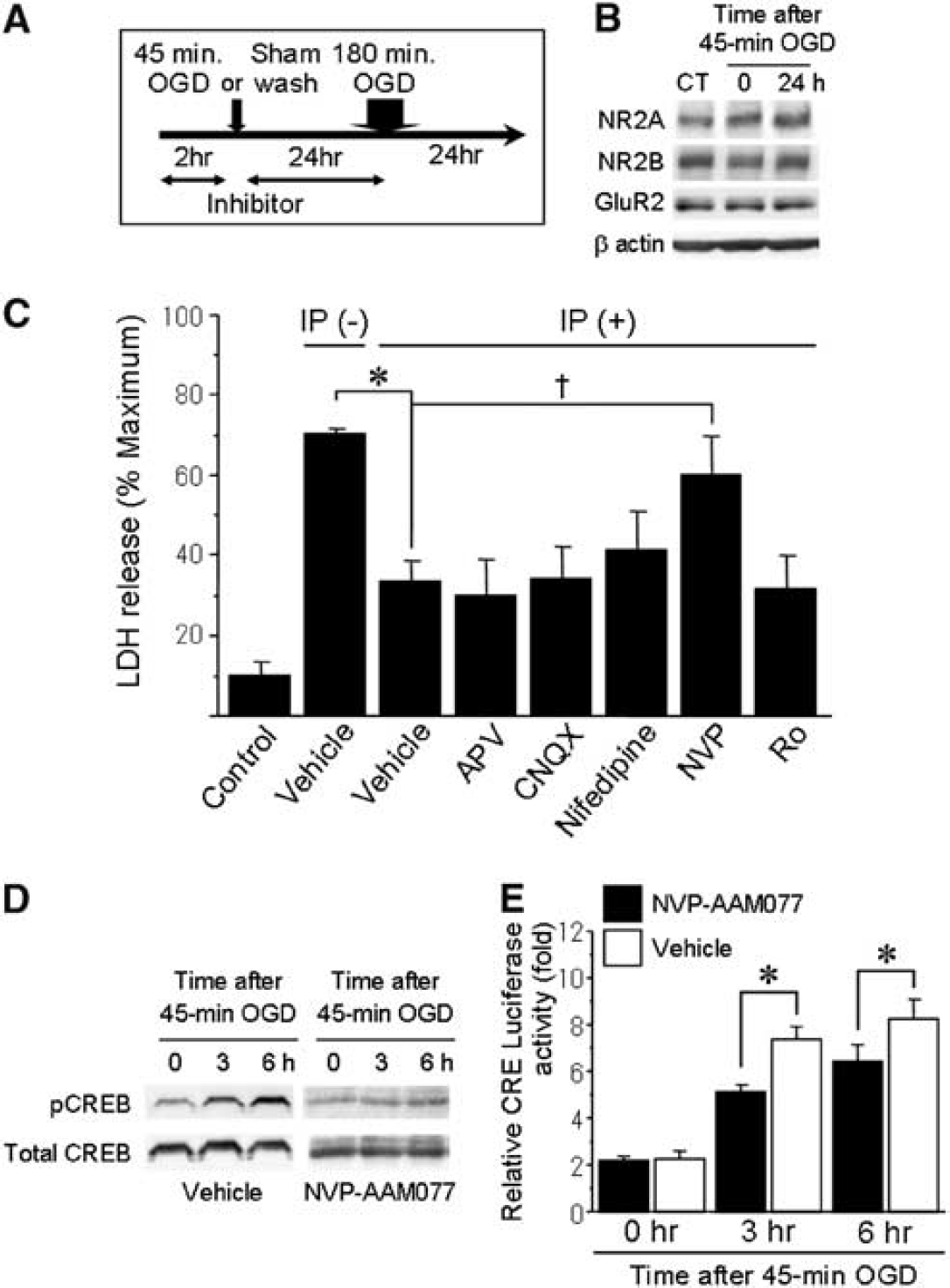
Effects of NVP-AAM077, a NR2A-specific antagonist, on ischemic tolerance and CRE transcriptional activity after sublethal OGD. (
These results show that inhibiting NR2A-containing NMDA receptors extinguishes ischemic tolerance with a correlated reduction of CRE activity. To determine whether activation of NR2A receptors would induce ischemic tolerance, we stimulated neurons by treatment with bicuculline and 4-AP, which has been shown earlier to stimulate neuronal synapses and activate NR2A receptors (Figure 5A) (Liu et al, 2007). As shown in Figure 5B, CRE activity levels significantly increased 3 and 6 h after administration of bicuculline and 4-AP, compared with vehicle-treated controls. Further, without preconditioning ischemia, ischemic tolerance was induced by pretreatment with bicuculline and 4-AP for 24 h before lethal ischemia (Figure 5C). To evaluate whether NVP-AAM077 blocked neuroprotective effect of bicuculline and 4-AP, we designed additional experiments; cells were treated with NVP-AAM077 or vehicle together with bicuculline and 4-AP for 24 h before lethal OGD (Figure 5D). The CRE activity levels significantly decreased 3 and 6 h after administration of NVP-AAM077 group compared with vehicle group (Figure 5E). Further, the neuroprotective effect of bicuculline and 4-AP against lethal OGD was attenuated by treatment with NVP-AAM077 (Figure 5F). These results suggest that activation of NR2A-containing NMDA receptors is sufficient to induce ischemic tolerance mediated by enhancement of CRE activity.
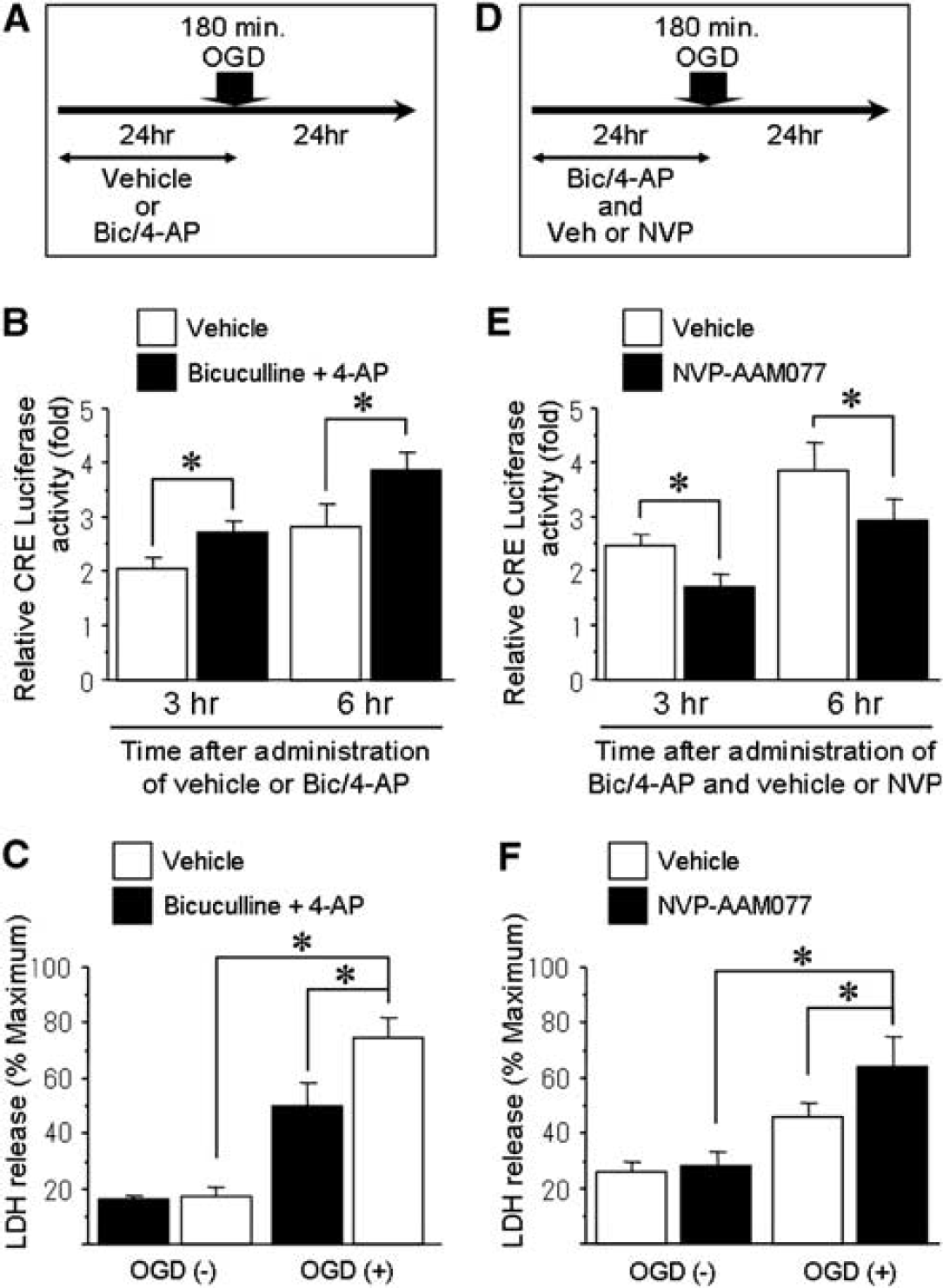
Induction of ischemic tolerance by stimulating neuronal synapses. (
NR2A Activation Enhances Brain-Derived Neurotrophic Factor Exon IV Promoter Activity Through Cyclic-AMP Response Element-Binding Protein Signaling
The CREB leads to neuroprotection mediated by transcription of downstream gene targets containing CRE motifs in their promoters; targets include BDNF, TrkB, and Bcl-2 (Deogracias et al, 2004; Riccio et al, 1999; Shieh et al, 1998). In particular, BDNF is a strong neurotrophic factor and TrkB, a receptor of BDNF also is a target for CREB regulation. Therefore, to elucidate the involvement of factors downstream of CREB, we investigated bdnf exon IV and mutant-bdnf (ΔCRE) exon IV transcriptional activity (Figure 6A). Promoter IV obviously contributes to activity-dependent BDNF transcription (Timmusk et al, 1993). More recently, promoter IV-driven BDNF transcription has been shown to have a critical function on synaptic plasticity with promoter IV mutant mice (Sakata et al, 2009). To dissect the function of the CRE cis-element within the bdnf promoter, we prepared a bdnf reporter gene construct with a mutation in the CRE-binding site. After sublethal OGD, BDNF levels significantly increased compared with basal levels, whereas mutant-BDNF levels were unaffected (Figure 6B). When NR2A was inhibited by NVP-AAM077, BDNF activity levels was significantly reduced compared with vehicle treatment (Figure 6B). In contrast, when the CRE was mutated, the inhibitory effect of NVP-AAM077 was not observed. The BDNF activity levels significantly increased 6 and 12 h after administration of bicuculline and 4-AP compared with mutant-BDNF activity (Figure 6C). These results suggest that BDNF transcriptional activity depends on CRE motifs and that a CREB cascade originated from NR2A enhances BDNF expression.
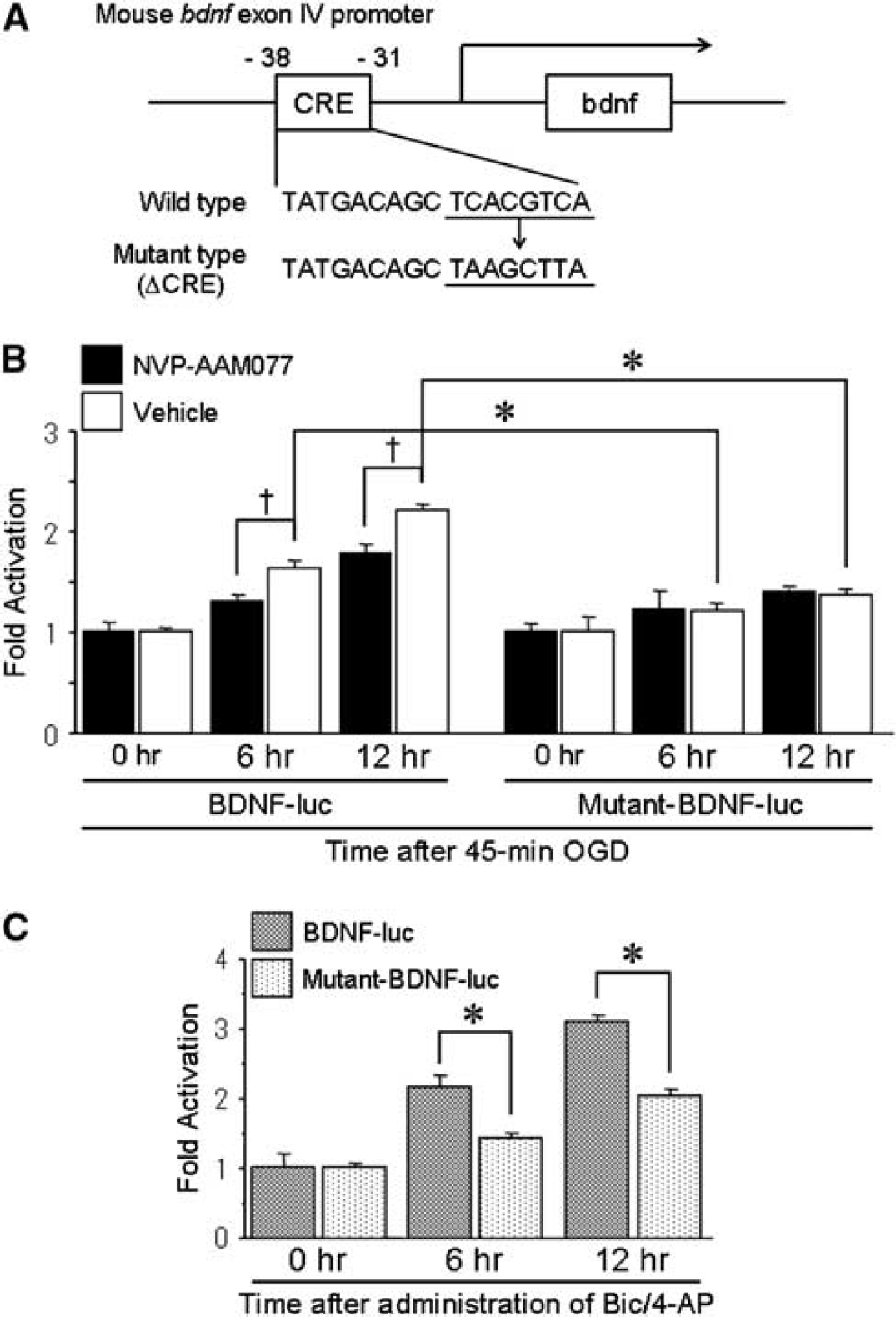
Increased BDNF transcriptional activity levels downstream of CREB after sublethal ischemia and synaptic stimulation. (
Discussion
In this study, we present two major evidences. First, CREB signaling originated from NR2A is an essential step in the induction of ischemic tolerance. In addition, sublethal ischemia induced cross-tolerance against otherwise lethal treatment with glutamate or hydrogen peroxide. Thus, this preconditioning stimulus may make neurons resistant to various insults related to excitotoxicity and oxidative stress. Second, preconditioning-induced neuroprotection was only attenuated by treatment with NVP-AAM077, but not other glutamate receptor antagonists. Activation of NR2A-containing NMDA receptors by administration of bicuculline and 4-AP increased CRE activity levels and led to neuroprotection. Taken together, these results suggest that CREB signaling originated from NR2A-containing NMDA receptors has a pivotal function in ischemic tolerance.
Considerations that should be taken into account when discussing stimulation of synapses with bicuculline and 4-AP include developmental and regional expression of NMDA subtypes and proportion of synaptic NMDA receptors containing the NR2B subunit (Monyer et al, 1994). NR2A-containing NMDA receptors are predominantly expressed at synapses; however, NR2B-containing receptors are thought to exist predominantly at extrasynaptic sites, but are also expressed at synapses (Massey et al, 2004). In addition, cortical neurons are thought to express NR1/NR2A/NR2B heterotrimeric NMDA receptors (Luo et al, 1997). As the function of NR1/NR2A/NR2B heterotrimeric receptors remains unclear, further studies to clarify their effects on neuronal survival or death and ischemic tolerance are necessary.
Tissue damage in ischemia is the result of a complex pathophysiological cascade, which comprises many distinct pathological stresses including hypoxic stress, oxidative stress, inflammatory stress, and glutamate excitotoxic stress. Ischemic tolerance protects the brain itself mediated by endogenous multiple pathways against these noxious stresses after ischemia (Dirnagl et al, 2009). One of the key regulators involved with ischemic tolerance is hypoxia-inducible factor. Ischemic tolerance activates hypoxia-inducible factor-1 and leads to neuroprotection via its target gene (Bernaudin et al, 2002). Also, ischemic tolerance attenuates oxidative stress by modification of nuclear factor κ B-driven gene and regulation of heat shock protein (Blondeau et al, 2001; Kirino et al, 1991). The preconditioning with lipopolysaccharide, a ligand of Toll-like receptors, stimulates antiinflammatory processes, suppresses proinflammatory pathways, and induces ischemic tolerance (Stenzel-Poore et al, 2007; Stevens and Stenzel-Poore, 2006). In addition, glutamate excitotoxicity is important in the pathophysiology of ischemic brain injury, which induces massive release of glutamate into extracellular space. Ischemic preconditioning might induce tolerance either by lowering excessive glutamate release or by increasing glutamate uptake (Romera et al, 2007). Activation of glutamate receptors causes calcium influx, activates calcium/CaM kinase, PI3K-Akt pathway, and Ras-MAPK cascade (Soriano et al, 2006; Wang et al, 2007), and in turn converge on CREB activation and upregulation of its target genes. Also, NR2A-containing synaptic NMDA receptors activate the extracellular signal-regulated kinase (ERK) pathway (Leveille et al, 2008). In fact, both Akt and ERK pathways not only activate CREB signaling but also mediate ischemic tolerance, and then, the target genes of CREB, such as Bcl-2, have a crucial function on ischemic tolerance via its antiapoptotic mechanism. Therefore, CREB signaling through NR2A could be one of the most important pathways involved in ischemic tolerance.
In excitotoxic conditions including ischemia, abnormal intracellular calcium entry triggers cell death through calcium overload (Bano and Nicotera, 2007). As shown recently, after an ischemic insult, other plasma membrane channels such as transient receptor potential (TRP) channel and acid sensing ion channel can contribute to calcium accumulation that induces neuronal death (Sun et al, 2009; Xiong et al, 2004). Also, earlier study reported that TRPC6, one of the TRP channel subfamilies, was associated with CaMKIV-CREB-dependent gene transcription including BDNF (Zhou et al, 2008). Thus, under ischemic condition, there is a possibility that calcium inflow routes besides NMDA receptors contribute to CREB pathway. Therefore, as shown in Figure 4E, transcriptional activity of CRE was not completely suppressed with NVP-AAM077.
An enormous number of studies in vitro and in vivo have suggested that NMDA receptor antagonists are effective in ischemic neuronal death. However, clinical stroke trials using NMDA antagonists have all failed. At least in part, narrow time windows for treatment may be important. Indeed, in the early phase of stroke, overactivation of NMDA receptors is detrimental, but in the delayed phase, NMDA signals may be required for recovery, including neuroplasticity and protection. Meanwhile, activation of NR2A-containing NMDA receptors may have some benefits compared with NMDA receptor antagonists that have been applied to stroke patients in previous trials. It remains possible that stimulating NR2A in clinical settings may attenuate neuronal injury and lead to a more favorable outcome than not stimulating NR2A. In addition to ischemic stroke, activation of NR2A-containing NMDA receptors may be a potential avenue to treat chronic neurodegenerative diseases (Ikonomidou and Turski, 2002).
Ischemic tolerance, an endogenous adaptive response, has received intense interest both clinically and nonclinically. Thus, we have clarified that stimulation of NR2A-containing NMDA receptors protect the brain against lethal ischemia mediated by neuroprotective signaling via CREB. This understanding of the mechanisms engaged in ischemic tolerance may allow novel therapeutic strategies for neuroprotection.
Footnotes
Acknowledgements
This work was supported by a Grant-in-Aid from the Ministry or Education, Science, and Culture in Japan. We thank Ms C Kurano for secretarial assistance and Ms K Nishiyama for laboratory assistance.
The authors declare no conflict of interest.