
Abstract
Alpha-synuclein oligomerization and aggregation are considered to have a role in the pathogenesis of neurodegenerative diseases. However, despite numerous
Keywords
Introduction
Alpha-synuclein is a protein of 140 amino acids that is predominantly expressed in presynaptic terminals throughout the central nervous system (Jakes et al, 1994; Maroteaux and Scheller, 1991). Under pathologic conditions, α-synuclein aggregates to form neuronal inclusions in several neurodegenerative diseases (Soto, 2003; Spillantini et al, 1997). Alpha-synuclein is the major component of Lewy bodies found in Parkinson's disease, dementia with Lewy bodies, and the Lewy body variant of Alzheimer's disease, as well as in glial cytoplasmic inclusions in multiple system atrophy (Baba et al, 1998; Spillantini et al, 1997; Tu et al, 1998; Ueda et al, 1993). Alpha-synuclein in these inclusions is reported to be nitrated, suggesting a role for oxidative and nitrative damage in α-synuclein modification and aggregation (Giasson et al, 2000). Fibril formation is facilitated by factors such as acidic pH and divalent metals.
To gain further insight whether α-synuclein aggregates formed during ischemia are toxic, we used transgenic mice expressing a human mutation (A30P) identified in a familial form of Parkinson's disease (Kruger et al, 1998). Expression in transgenic mice of human synuclein mutations A53T or A30P induces abnormal cellular accumulations in the central nervous system (Kahle et al, 2000; Masliah et al, 2000; van der Putten et al, 2000).
Materials and methods
Proximal Middle Cerebral Artery Occlusion
Animal housing, care, and application of experimental procedures were carried in accordance with the regulations of the Ethics Committee of Hacettepe University (2002/44-2). In the first part of experiments, Swiss Albino mice (weighing 35 to 40 g) were anesthetized with chloral hydrate (400 mg/kg intraperitoneal), and proximal occlusion of the right MCA was performed using a nylon filament as described previously (Gursoy-Ozdemir et al, 2000) to investigate α-synuclein aggregation in wild-type animals. In brief, a silicon-coated nylon filament (8/0) was inserted into the right common carotid artery through a small incision proximal to the bifurcation and advanced into the internal carotid artery up to the origin of the MCA (10 mm from the bifurcation). Mice received 10 IU of heparin before ischemia, and blood pressure was monitored using a catheter placed in the common carotid artery. Regional cerebral blood flow (rCBF) was monitored by laser-Doppler flowmetry (Periflux PF 2B, Perimed, Jarfalla, Sweden). After obtaining a stable 10-minute epoch of preischemic rCBF, the MCA was occluded and rCBF was continuously monitored during ischemia and the first 20 minutes of reperfusion. Reperfusion was accomplished by pulling the filament back. After 30 minutes of MCA occlusion and 72 hours of reperfusion, animals were anesthetized with a high dose (1 g/kg) of chloral hydrate and cardiovascularly perfused with 4% formaldehyde. Mice were decapitated and their brains were either cryoprotected or paraffin embedded.
Proximal and Distal Middle Cerebral Artery Ischemia in Transgenic Mice
For studying the impact of transgenic α-synuclein on brain infarct, 8-week-old [
In another set of transgenic and wild-type C57BL/6 mice, SNN was induced by compressing the distal MCA for 20 minutes with a glass pipette inserted through a cranial window using a micromanipulator without opening the duramater. The pipette tip was thinned to 50 to 70 μm and blunted. The success of the compression was verified by observing a decrease in rCBF to ischemic values in the distal MCA territory with laser-Doppler flowmetry. After this brief ischemia, 72 hours of reperfusion was required for adequate number of SNNs to appear. At the end of reperfusion, mice were perfused with 4% paraformaldehyde under ketamine anesthesia. To avoid dark neurons in SNN experiments, the decapitated head was kept in 4% paraformaldehyde for 12 hours before the brain was taken out of the skull. The brains were then fixed in 4% paraformaldehyde for an additional 12 hours.
In these mice, blood pressure was monitored noninvasively (NIBP Controller, AD Instruments Pty Ltd., Bella Vista, NSW, Australia) from the tail.
Evaluation of Ischemia Outcome
Infarct volumes and the number of selectively necrotic neurons were detected on hematoxylin and eosin-stained 5-μm-thick brain sections. The infarct area was measured using image-analysis software (NIH Image; http://rsb.info.nih.gov/nih-image/) on the posterior surface of each section, and infarct volumes were calculated by summing the areas of sequential 2-mm-thick sections.
For evaluation of SNN, the number of neurons showing necrotic changes such as pyknosis, karyorhexis, karyolysis, and eosinophilia was counted on 6 microscopic fields randomly selected from the distal MCA territory at × 1,000. Neurons were identified by light microscopic criteria (a large cell body, a large nucleus with a single, prominent, centrally located nucleolus) because the experiments performed to develop the SNN model (Arsava et al, 2009) unambiguously showed that degenerating cells were NeuN-positive neurons.
Cells with apoptotic nuclei were counted manually under dark-field microscopy (Lange et al, 1999) at × 400 magnification in 3 selected brain regions (namely, the frontal cortex, the parietal cortex, and the lateral striatum) in the proximal MCA occlusion model and, in 3 adjacent microscopic fields in the frontoparietal cortex in the distal occlusion model, respectively. To reliably detect cells with apoptotic nuclei, dark and bright field images of the same area were captured and superimposed.
Mean values of infarct volumes, number of SNNs, bright nuclei in dark field and immunopositive neurons were compared with Mann—Whitney
Immunohistochemistry
Coronal brain sections were stained with anti-α-synuclein (sheep polyclonal antibody-AB5334P, Chemicon, Temecula, CA, USA), β-synuclein (rabbit polyclonal antibody-6485 (Kahle et al, 2000), ubiquitin (rabbit polyclonal antibody, RDI-UBIQUITabR, Research Diagnostics, Concord, MA, USA), antinitrotyrosine (anti-NT) (rabbit immunoaffinity purified IgG-06-284, Upstate, Lake Placid, NY, USA), caspase-3-p20 (rabbit polyclonal antibody-9661S, Cell Signaling, Beverly, MA, USA) or glial fibrillary acidic protein (mouse monoclonal, Sigma, St Louis, MO, USA) antibodies.
Fifteen to 20-μm-thick frozen sections were washed in phosphate-buffered saline and permeabilized with 0.2% Triton-X. The tissues were then incubated with 3% bovine serum albumin and 10% host serum of the secondary antibody to eliminate nonspecific binding. The sections were incubated with primary antibodies at 1:100 (synucleins) or 1:500 (ubiquitin) dilution for 12 hours at +4°C and then with secondary antibodies (1:100 dilution, Jackson ImmunoResearch, West Grove, PA, USA) for 1 hour at 37°C. Sections were mounted with 50% glycerol. The immunolabeled frozen sections were examined under a Nikon Eclipse E600 upright microscope (Nikon, Tokyo, Japan) using appropriate filter sets and the images were captured through a TV lens (
Paraffin-embedded sections of 5-μm thickness were deparaffinized at 56°C overnight and rehydrated by graded alcohol. Anti-3-NT and caspase-3-p20 antibodies were diluted (1:200) in phosphate-buffered saline. For staining with antiubiquitin antibody, endogen peroxidase activation was blocked with H2O2, and sections were then microwaved in 1 mmol/L EDTA buffer for antigen retrieval. Antiubiquitin antibody was diluted (1:500) in 3% bovine serum albumin. After incubation with primary antibodies (ubiquitin for 2 hours and the others, overnight) serial sections were stained with conventional avidin—biotin—peroxidase technique and diaminobenzidine was used as the chromogen. Hematoxylin was used as the counterstain. Sections were examined under bright-field microscope (Nikon Eclipse E600, Nikon) and images were captured and analyzed as described above.
The number of mice used for immunohistochemistry studies is given in parentheses in the ‘Results’ section. Several coronal brain sections were studied from each mouse.
Paraffin-Embedded Tissue Blotting
To test whether anti-α-synuclein-immunostained aggregates were proteinase-K resistant, paraffin-embedded tissue blotting method (Kramer and Schulz-Schaeffer, 2007; Neumann et al, 2002; Schulz-Schaeffer, 2002) was used. Paraffin-embedded 5-μm-thick sections passing through the anterior commissure were deparaffinized at 56°C overnight, and rehydrated by graded alcohol and xylol. Dried tissue sections layered on nitrocellulose membranes were prewetted and digested with 0.2 mg/mL proteinase-K (Chemicon) for 8 hours at 55°C. After washing, the membranes were treated for 10 minutes with 3 mol/L guanidine isothiocyanate in 10 mmol/L Tris-HCl, pH 7.8, for optimal epitope retrieval. The tissues were then incubated with anti-α-synuclein (1:200, overnight, 4°C, sheep polyclonal antibody-AB5334P, Chemicon), and immunodetection of α-synuclein aggregates was performed using the conventional streptavidin—horseradish peroxidase technique (Dako cytomation kit, Glostrup, Denmark). Diaminobenzidine was used as the chromogen. Proteinase-K step was omitted for conventional α-synuclein immunohistochemistry of simultaneously processed control paraffin sections.
Thioflavin-S Staining
To test whether anti-α-synuclein immunostained accumulations were protein aggregates, we used thioflavin-S (TS-T1892, Sigma) staining. Deparaffinized and rehydrated sections were incubated for 8 minutes with filtered 1% thioflavin-S in 50% ethanol in the dark at room temperature. Sections were then washed under light protection with 80% and 95% ethanol and then with double-distilled water 3 times for 1 minute to remove ethanol. The sections were finally incubated for 3 minutes in phosphate-buffered saline under light protection and mounted with DPX (4458, Sigma).
Detection of Insoluble Protein Aggregates with Western Blotting
Mice subjected to 30 minutes MCA ischemia and 72 hours of reperfusion (
Results
Ischemia Induces Insoluble Alpha-Synuclein Aggregates
Western Blotting
We observed significant α-synuclein oligomerization in western blots prepared from the ischemic MCA territory of wild-type mice subjected to 30 minutes of ischemia and 72 hours of reperfusion (
Immunohistochemistry
We also searched for α-synuclein aggregates in the ischemic brain by immunohistochemistry to detect their cellular distribution. We used an antibody developed against human α-synuclein because it shows high affinity to aggregated α-synuclein (e.g., to Lewy bodies) (Jakes et al, 1999). In line with previous reports (Kahle et al, 2000, 2001), we observed a diffuse, neuropillic α-synuclein staining in the nonischemic brain (
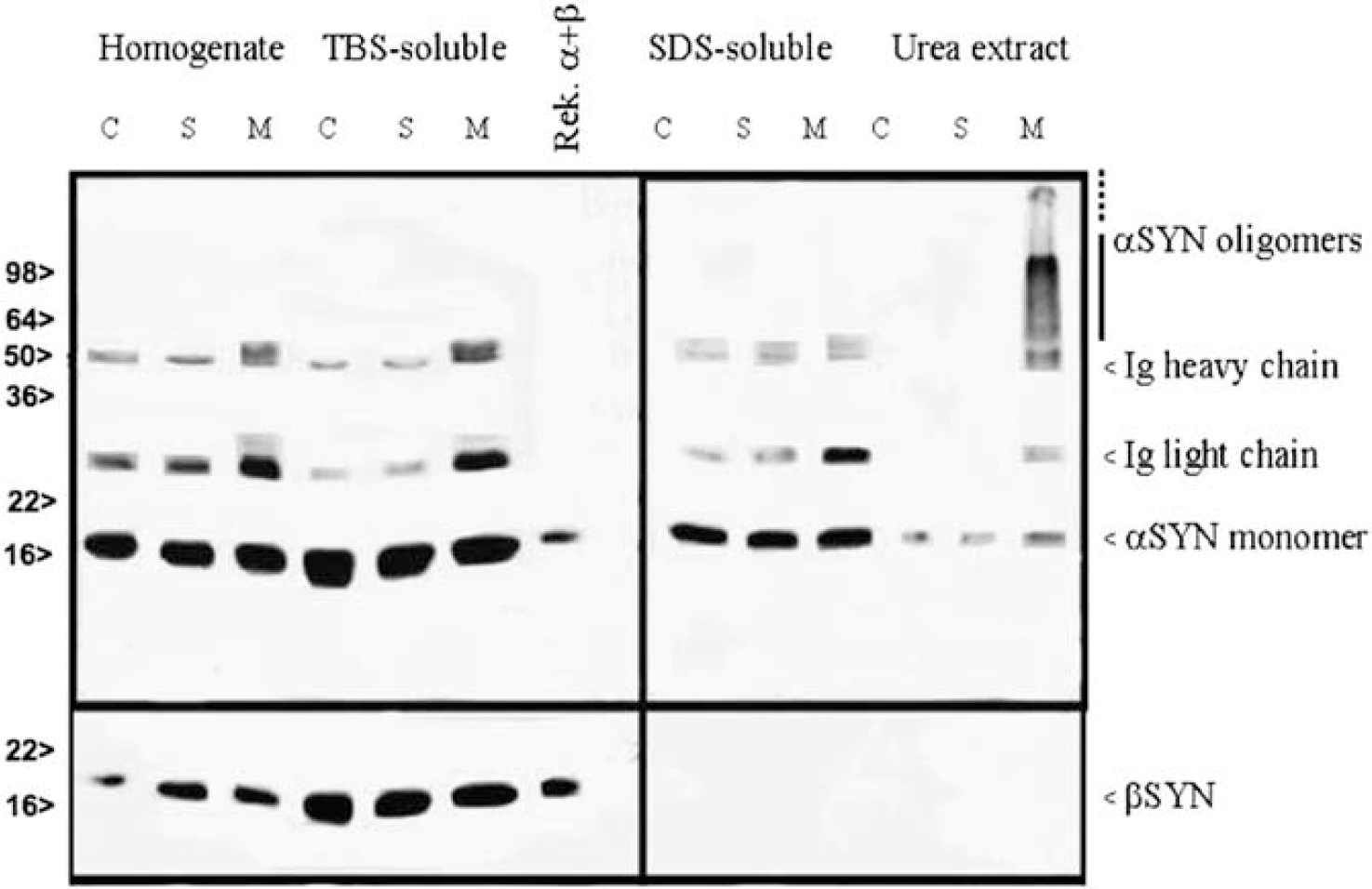
Detergent-insoluble α-synuclein monomers and oligomers were detected in the ischemic MCA area (M) but not in the nonischemic contralateral hemisphere (C) or in the sham-operated brains (S). Beta-synuclein bands were found in the soluble fraction but no bands or aggregates were detected in the urea extract of the ischemic MCA area. MCA, middle cerebral artery.
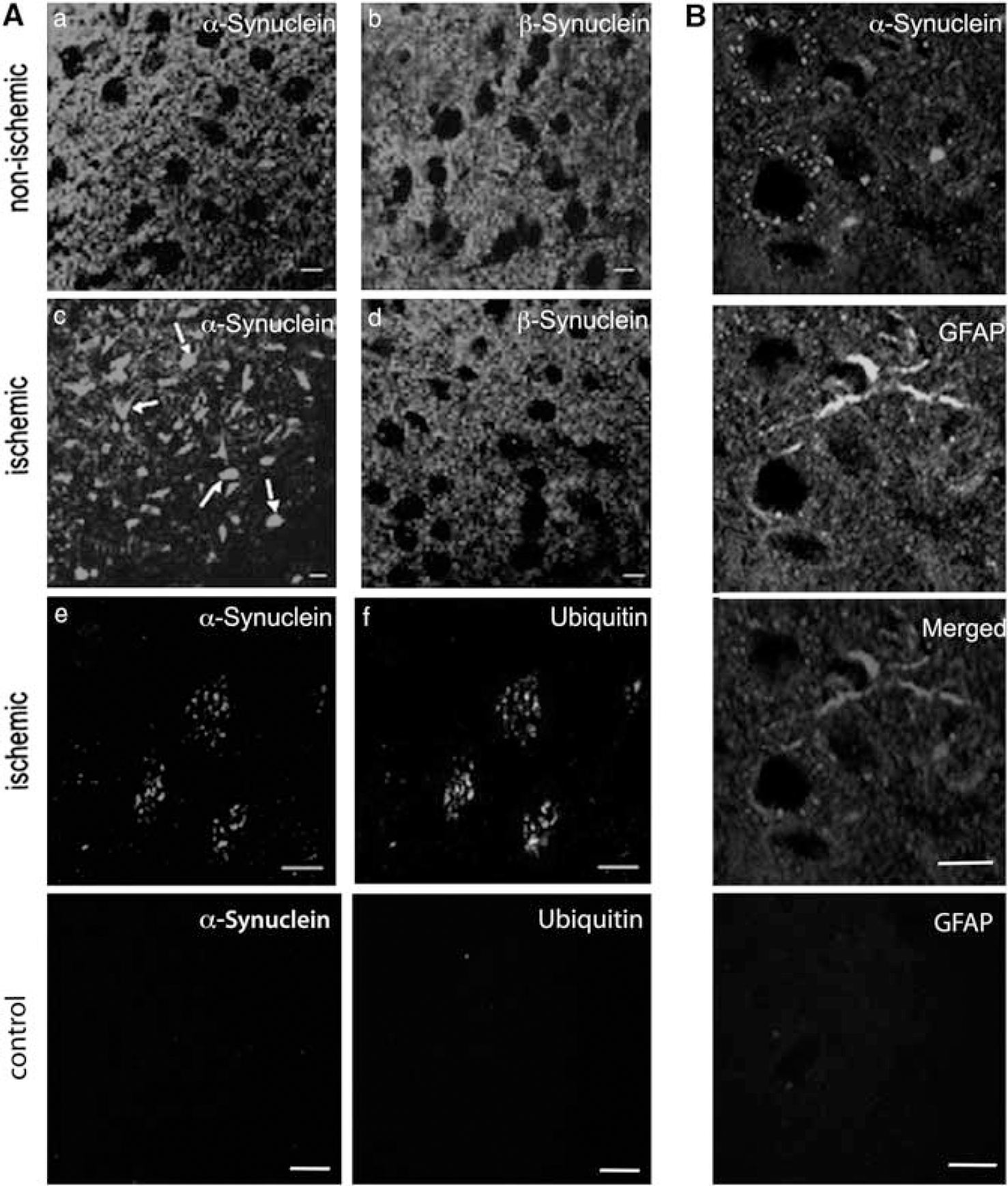
Ischemia induces neuronal α- but not β-synuclein aggregation. (
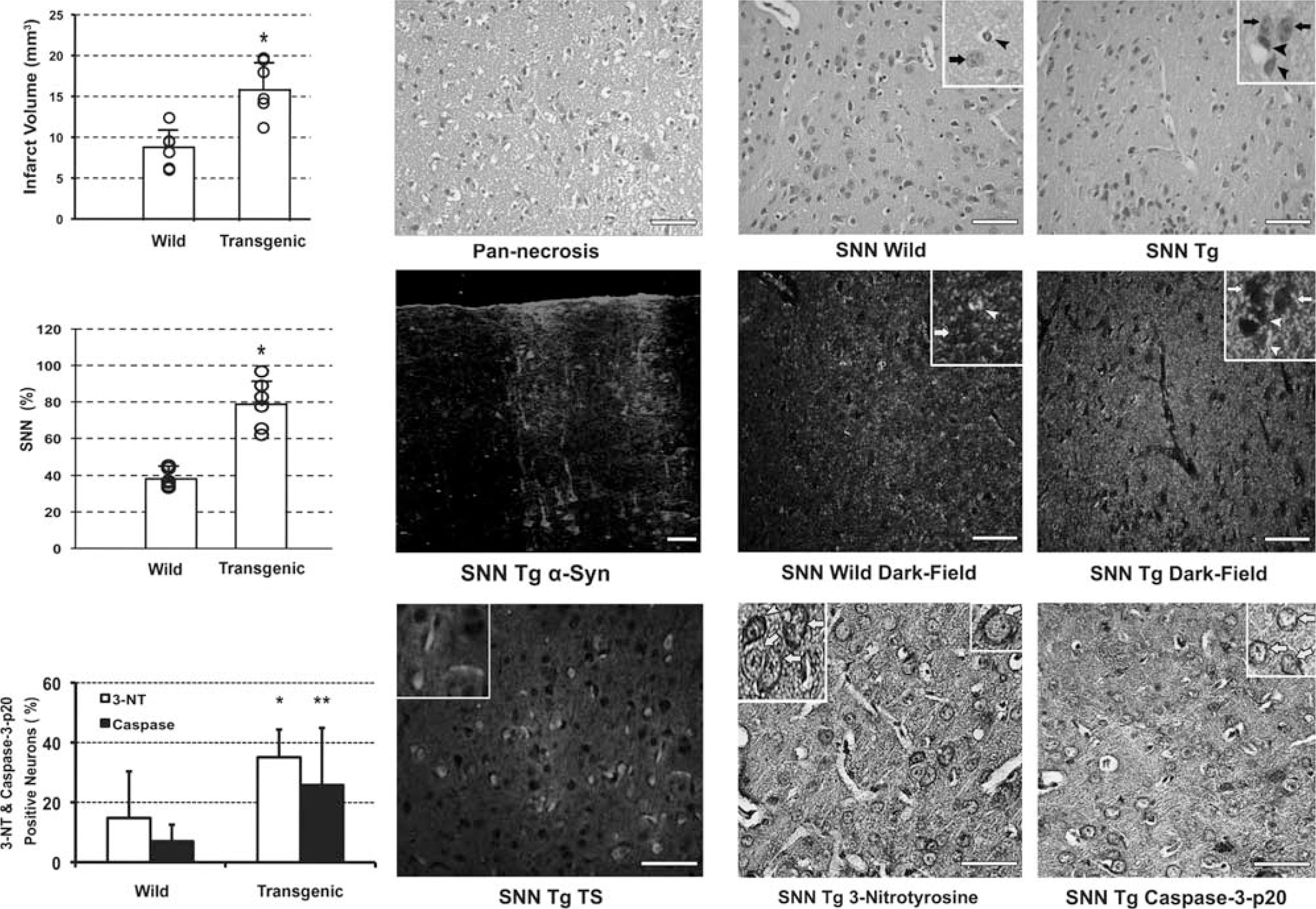
[A30P]alpha-synuclein transgenic C57BL/6 mice developed larger lesions compared with wild-type C57BL/6 mice when subjected to transient ischemia. Thirty-minutes proximal MCA occlusion (upper row) caused pannecrosis (microphotograph at × 400, H&E staining). Transgenic mice had significantly larger infarct volumes (graph, ∗ denotes
We also stained the sections with β-synuclein to compare its staining pattern in the ischemic hemisphere with that of α-synuclein (
Aggregates Colocalize with Ubiquitin
Alpha-synuclein aggregates are ubiquitinated in inclusion bodies seen in several neurodegenerative diseases (Goedert, 2001; Matsuzaki et al, 2004; Soto, 2003; Spillantini et al, 1997, 1998). We therefore examined colocalization of the α-synuclein aggregates with ubiquitin immunoreactivity in ischemic brain sections. The diffuse ubiquitin immunoreactivity observed in nonischemic cells was replaced by a coarse granular pattern after ischemia—reperfusion on paraffin-embedded brain sections (
Alpha-Synuclein Aggregates are Proteinase Resistant
The paraffin-embedded tissue blotting method (Kramer and Schulz-Schaeffer, 2007; Neumann et al, 2002; Schulz-Schaeffer, 2002) allows selective detection of protein aggregates because nonaggregated proteins are readily degraded by proteinase-K treatment. In untreated paraffin-embedded sections, we detected a diffuse neuropilic α-synuclein immunostaining in the nonischemic hemisphere, whereas somatic α-synuclein clumps were seen in ischemic neurons similar to our observations on frozen sections. After proteinase-K treatment, neuropilic α-synuclein immunoreactivity in the nonischemic areas completely disappeared, whereas somatic α-synuclein clumps and part of the neuropilic immunostaining in the ischemic MCA area persisted (
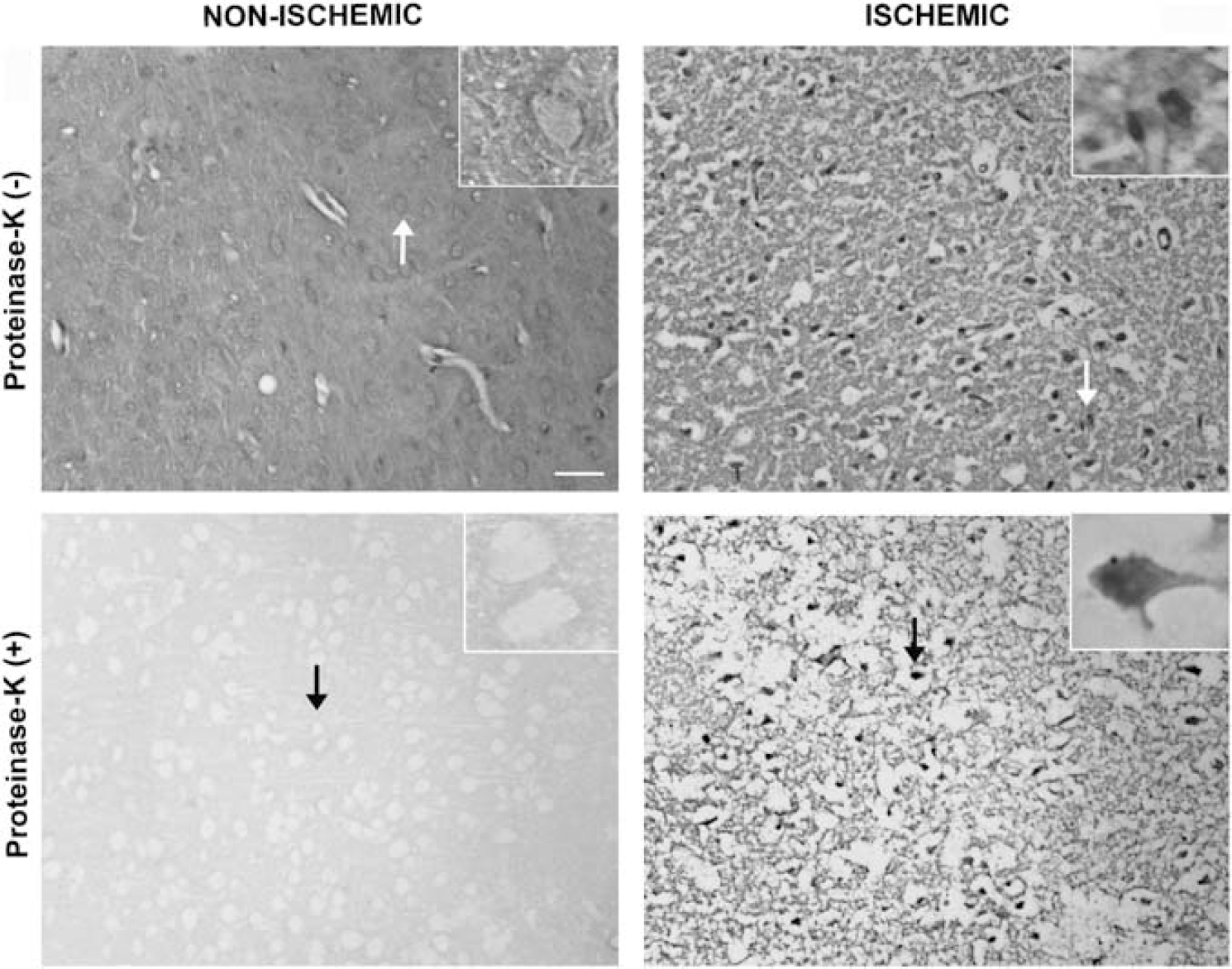
Paraffin-embedded tissue (PET) blotting method illustrated that α-synuclein clumps observed with immunohistochemistry were proteinase-K resistant aggregates. In sections untreated with proteinase-K (upper row), a diffuse neuropilic α-synuclein immunostaining was detected in the nonischemic hemisphere, whereas somatic α-synuclein clumps were seen in neurons in the ischemic area 72 hours after 30 minutes MCA occlusion. After proteinase-K treatment, neuropilic α-synuclein immunoreactivity in the nonischemic areas completely disappeared, whereas somatic α-synuclein clumps and part of the neuropilic immunostaining in the ischemic MCA area persisted (lower row). Ischemia-induced neuropilic sponginess looks exaggerated than expected for this time point because of proteinase-K-induced tissue degradation and PET blotting procedures. Microphotographs were taken from the frontoparietal cortex and insets are enlarged views of the areas marked with arrows. Scale bar: 20 μm. MCA, middle cerebral artery.
Alpha-Synuclein Transgenic Mice are More Sensitive to Cerebral Ischemia
We subjected wild-type and [A30P]α-synuclein transgenic C57BL/6 mice to 30 minutes proximal MCA occlusion or to 20 minutes distal MCA compression to compare their vulnerability with ischemic brain injury. We chose the 30-minute ischemia plus 24-hour reperfusion model because it produces a fairly consistent infarct size providing satisfactory statistical power with a small number of animals (
To investigate whether ischemic cell death mechanisms were modified in transgenic mice, we counted the nuclei with apoptotic morphology under dark-field microscopy, in which clumped chromatin refracts light and appears as bright objects in a dark background, whereas dispersed chromatin in necrotic cells does not (Lange et al, 1999). We found that transgenic mice had a similar number of apoptotic nuclei compared with wild types in both models; 1,181±589/mm2 versus 1,380±397/mm2 in the proximal and 970±568/mm2 versus 935±297/mm2 in SNN models (
There were no significant differences between the transgenic and wild-type groups regarding the vasculature at macroscopic and microscopic levels, for rCBF recorded during ischemia and the first 20 minutes of reperfusion, as well as for arterial blood pressure (Table 1), suggesting that the observed differences between the groups were not caused by hemodynamic changes but by an increased sensitivity to ischemic injury in transgenic mice.
Physiologic parameters
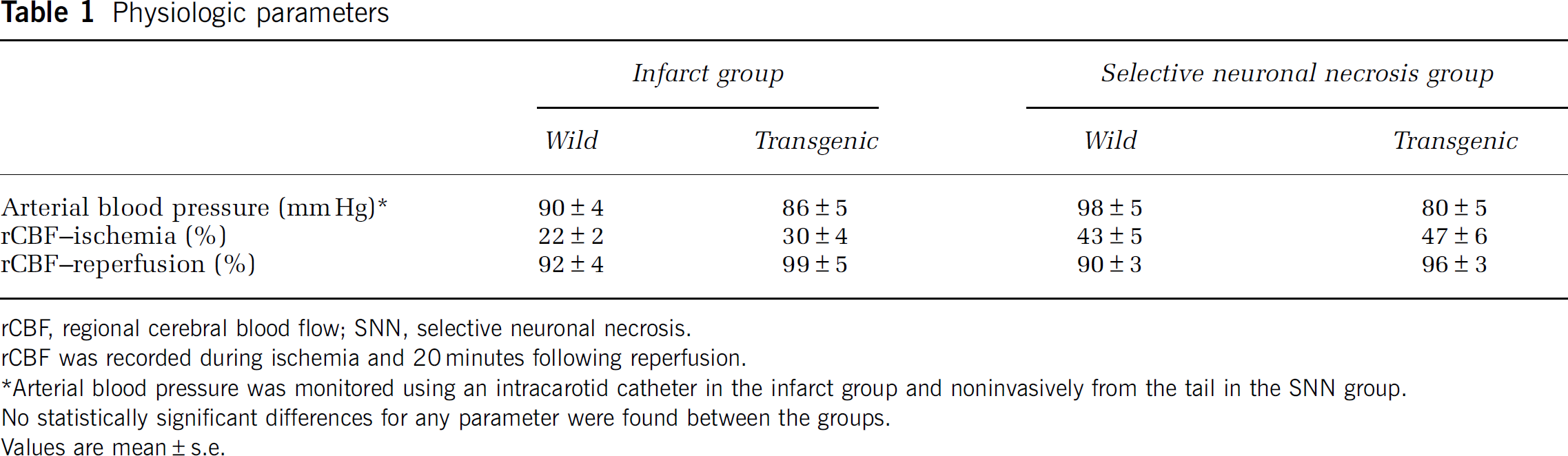
rCBF, regional cerebral blood flow; SNN, selective neuronal necrosis.
rCBF was recorded during ischemia and 20 minutes following reperfusion.
Arterial blood pressure was monitored using an intracarotid catheter in the infarct group and noninvasively from the tail in the SNN group.
No statistically significant differences for any parameter were found between the groups.
Values are mean±s.e.
Discussion
This study shows that transient MCA ischemia induces oligomerization of wild-type α-synuclein in the mouse brain. This effect is isoform specific because β-synuclein, the isoform known to be resistant to fibril formation (Giasson et al, 2001; Kahle et al, 2001), does not form oligomers. Immunohistochemistry illustrated that α-synuclein aggregates in neurons were proteinase-K resistant and colocalized with ubiquitin immunoreactivity as seen in synucleinopathies (Goedert, 2001; Matsuzaki et al, 2004; Soto, 2003; Spillantini et al, 1997, 1998). Importantly, our findings also showed that α-synuclein aggregates not only form during ischemia but also aggravates oxidative/nitrative stress and negatively impact neuronal survival in transgenic mice expressing the human α-synuclein with the [A30P] mutation, supporting the idea that α-synuclein misfolding may be neurotoxic.
Western blots convincingly showed that ischemic wild-type brain contained an abundance of insoluble oligomers of α-synuclein (Kahle et al, 2001; Miake et al, 2002; Neumann et al, 2002). Alpha-synuclein-immunopositive clumps observed with immunohistochemistry, very likely represented the aggregated oligomers because they were resistant to proteinase-K treatment and were also immunopositive for ubiquitin (Goedert, 2001; Matsuzaki et al, 2004; Soto, 2003; Spillantini et al, 1997, 1998). Ischemia may have disrupted axonal transport of α-synuclein and led to its somatic accumulation and predisposed to aggregation in the cytoplasm in addition to the proteinase-K-resistant aggregation in the neuropil. Somal accumulation of α-synuclein was also found in mice subjected to a chronic 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine regimen (Vila et al, 2000). Once oligomerization is triggered during ischemia, initial aggregates may continue to trigger further oligomerization despite reperfusion (Soto, 2003; Wood et al, 1999). Further strengthening the specificity of these observations, the above findings were not observed with β-synuclein, which is not amyloidogenic (Giasson et al, 2001; Kahle et al, 2001).
Aggregates containing ubiquitinated proteins have been reported to form in neurons after experimental cerebral ischemia; Hu et al (2000) showed that transient global cerebral ischemia caused protein aggregation in hippocampal CA1 neurons. Two hours of MCA occlusion was also reported to induce protein aggregation in neocortical neurons (Hu et al, 2001). After cerebral hypoxia—ischemia, misfolded proteins were shown to accumulate in the lumen of the endoplasmic reticulum (DeGracia and Montie, 2004). This accumulation leads to stress responses, inhibition of protein synthesis, and activation of programmed cell death through the mitochondrial apoptotic pathway (Paschen and Mengesdorf, 2005).
Similarly, the α-synuclein oligomers and aggregates that we observed in ischemic neurons may unfavorably impact neuronal survival as suggested by larger lesions seen in A30P transgenic mice subjected to focal cerebral ischemia. A30P α-synuclein has a higher tendency to oligomerize when exposed to oxidizing conditions
There is still controversy whether inclusion bodies that are considered pathologic hallmarks of various neurodegenerative diseases, are toxic to the affected neurons (Arrasate et al, 2004; Kopito, 2000; Kramer and Schulz-Schaeffer, 2007; Ross and Poirier, 2005; Soto, 2003). In a recent postmortem study, of the 106 brains having α-synuclein-positive inclusions, only 30% had been clinically diagnosed as having a neurodegenerative disorder, whereas 30% did not have any neurologic impairment (Parkkinen et al, 2005). In fact, some experimental data imply that these inclusions may be formed as a defense against cellular stress induced by misfolded proteins (Arrasate et al, 2004; Giasson et al, 2000; Lee et al, 2001). These conflicting observations may be reconciled by assuming that clinical syndromes may result from a ‘critical mass’ of aggregated insoluble material, whereas lower amounts do not exert toxicity and may even be protective (Burn, 2006). Our findings from A30P transgenic mice strongly support the idea that misfolded α-synuclein can be neurotoxic in ischemic neurons. In an acute and severe insult such as ischemia, the threshold may be reached in a short time and toxicity induced by α-synuclein aggregates may contribute to cell death. Interestingly, in the above-mentioned postmortem study, more than one-third of patients with α-synuclein inclusions had died of primarily cerebrovascular and cardiovascular diseases, increasing the possibility that the ischemic/anoxic episodes to the brain may have promoted α-synuclein-positive inclusions as we observed in the mouse brain. Hence, further postmortem examination of human brains that had experienced brief ischemic episodes seems warranted to see whether they bore presynaptic α-synuclein aggregates in the absence of Lewy bodies (Kramer and Schulz-Schaeffer, 2007).
The term ‘selective neuronal necrosis (SNN)’ denotes the death of neurons with sparing of glial and vascular elements following mild and/or short-term ischemia. The process is different from pannecrosis, which affects all elements of the nervous system. Selective neuronal necrosis is assumed to have a role after transient ischemic attacks, and in the formation of silent brain lesions and vascular dementia (Vermeer et al, 2003). [A30T] transgenic mice developed more extensive SNN after brief, focal ischemia, suggesting that protein misfolding may have a role in the pathogenesis of silent brain lesions and the consequent vascular dementia as recently reported after traumatic brain injury in humans (Uryu et al, 2007). The SNN model we used appears promising to study the pathophysiology of aggregated or fibrillar α-synuclein in future studies because it achieves rapid aggregation
In conclusion, brief, transient ischemia induces oligomerization of wild-type α-synuclein. Alpha-synuclein aggregation is toxic to ischemic neurons. The A30P mutation promotes α-synuclein oligomerization and, hence, unfavorably affects neuronal survival when these mice are subjected to brief focal cerebral ischemia. The SNN model used in this study appears as a promising simple technique to investigate neuronal α-synuclein aggregation under
Footnotes
Acknowledgements
We thank Dr E Murat Arsava for the establishment of SNN method and, Elvan Ciftci and Ahmet Gürcan for their help with cell counting. Dr Turgay Dalkara's work is supported by the Turkish Academy of Sciences.
The authors declare no conflict of interest.