
Abstract
In this article, we examined theoretically the role of human cerebral glycogen in buffering the metabolic requirement of a 360-second brain stimulation, expanding our previous modeling study of neurometabolic coupling. We found that glycogen synthesis and degradation affects the relative amount of glucose taken up by neurons versus astrocytes. Under conditions of 175:115 mmol/L (∼1.5:1) neuronal versus astrocytic activation-induced Na+ influx ratio, ∼12% of astrocytic glycogen is mobilized. This results in the rapid increase of intracellular glucose-6-phosphate level on stimulation and nearly 40% mean decrease of glucose flow through hexokinase (HK) in astrocytes via product inhibition. The suppression of astrocytic glucose phosphorylation, in turn, favors the channeling of glucose from interstitium to nearby activated neurons, without a critical effect on the concurrent intercellular lactate trafficking. Under conditions of increased neuronal versus astrocytic activation-induced Na+ influx ratio to 190:65 mmol/L (∼3:1), glycogen is not significantly degraded and blood glucose is primarily taken up by neurons. These results support a role for astrocytic glycogen in preserving extracellular glucose for neuronal utilization, rather than providing lactate to neurons as is commonly accepted by the current ‘thinking paradigm’. This might be critical in subcellular domains during functional conditions associated with fast energetic demands.
Keywords
Introduction
Cerebral energetics is believed to be sustained via the complete oxidation of glucose (Bak et al, 2009). Nonetheless, regional glucose and oxygen utilization during focal brain stimulation exhibit a greater increase in glycolysis compared with oxidative metabolism, which is evidenced by a modest accumulation of lactate even in the presence of adequate oxygen tension in tissue (reviewed by Mangia et al, 2009a). Recently, it has been largely appreciated that the recruitment of brain cells in food-for-thought is not restricted to the neuronal electrical activity. Indeed, astrocytes are mandatory partners of neurons, and their metabolism is closely linked to synaptic activity (Hertz et al, 2007). The involvement of astrocytes in the coupling between glutamatergic neurotransmission and glucose utilization has posed into question the relative amount of neuronal versus astrocytic glucose uptake (Chih et al, 2001), and is at the basis of the astrocyte–neuron lactate shuttle hypothesis, which places astrocyte-derived lactate as a critical neuronal energy substrate (Pellerin and Magistretti, 1994). Conforming with this scenario, it has been suggested that energy transfer in the form of lactate from astrocytes to neurons is partly provided by glycogen metabolism (Brown, 2004).
In this study, we investigated the effect of glycogenolysis in buffering the high metabolic needs of the stimulated brain tissue by incorporating glycogen metabolism in mathematical models coupling brain electrical activity, metabolism, hemodynamics, and nutrients transport (Aubert and Costalat, 2005; Simpson et al, 2007; Mangia et al, 2009b). We specifically extended our previous integrative theoretical account (DiNuzzo et al, 2010), with a set of kinetic equations linking glycolysis to glycogen synthesis and breakdown in astrocytes. The main goal of this study was to evaluate the changes in cellular substrate availability, taking place when a fraction of the energy required by brain activation is supplied by stored carbohydrate. We found that the astrocytic support to neuronal function due to glycogen mobilization is identified by the indirect provision of glucose, not lactate to neurons (Swanson, 1992), contrary to the intuitive idea that an intermediary compound must be used by neurons for glycogen localization in astrocytes to be beneficial (Brown and Ransom, 2007). Specifically, glycogenolysis reduces astrocytic need for blood-borne interstitial glucose, thereby allowing its increased uptake into neurons. Our results are in agreement with the suggestion that, although the use of astrocytic glycogen is difficult to detect
Materials and methods
Kinetic modeling of cerebral glycogen metabolism was performed on top of several mathematical models of brain functional energetics (Aubert and Costalat, 2005; Simpson et al, 2007). In particular, we included a basic theoretical account for glycogen synthesis and mobilization to the key cell metabolic pathways of our previous integrative biochemical model (DiNuzzo et al, 2010). The model describes the time course of the main electrophysiological, metabolic, and vascular variables involved in neural activation. Briefly, nutrients traverse the blood–brain barrier and balance in cellular (neuron and astrocyte) and extracellular domains through diffusion or facilitative transport systems. Resting cell energy metabolism is regulated by basal energy consumption due to housekeeping processes plus activity of Na+/K+-ATPase after sodium leakage. Stimulation produces a rise in cerebral blood flow as well as inward Na+ currents in both neurons and astrocytes, which upregulate buffering of adenosine triphosphate (ATP) by adenylate and creatine kinases, and catabolism of glucose and lactate via glycolysis and respiration. We conformed to previous modeling works (Aubert and Costalat, 2005; DiNuzzo et al, 2010) by recasting all ATP-consuming processes as the gross cellular Na+ influx. Within astrocytes, in addition to Na+-glutamate cotransport, the modeled Na+ influx includes the effects of K+ sequestration via activation of Na+/K+-ATPase. Importantly, K+ sequestration is specifically relevant to the activation of glycogenolysis, as experimentally demonstrated in cell cultures (Dienel and Cruz, 2006, and references therein).
In the present extension of the model, we take into account brain glycogen storage and utilization during resting and stimulated conditions (Table 1). Glycogen is primarily located in astrocytes, so are the cellular enzymes that metabolize glycogen (Pfeiffer-Guglielmi et al, 2003). Therefore, we assumed that glycogen is entirely confined to astrocytes, with an equilibrium concentration of 14 mmol/L. Given an astrocytic element volume fraction of 0.25, this corresponds to a cerebral glycogen content of nearly 3.5 μmol/g, which is approximately the experimental value for the human brain (Oz et al, 2007), albeit somewhat lower compared with that found in rodent brain, where glucosyl units concentration has been found to range from 3 to 12 μmol/g depending on the extraction method and handling of the animal (Lowry et al, 1967; Cruz and Dienel, 2002). We ignored the subcellular distribution of glycogen in astrocytes, which can result in local concentration of up to 35 μmol/g in peripheral astrocytic processes (about 40% of astrocyte volume) (Hertz et al, 2007). It is noted that there is significant diurnal variation in total brain glycogen content, and the physiologic (activation or stress) as well as nutritional history of individual subjects may be important in defining the resting glycogen levels (Cruz and Dienel, 2002). We neglected the influence of organization and size of the glycogen particles on their metabolism. Because of the small changes in the glycogen level resulting from our simulations, we further assumed that the effective concentration of glucosyl units at glycogen outer branches is not considerably altered as glycogen size is reduced (Roach, 2002).
Model kinetic equations linking glycolysis to glycogen metabolism*

AMP, adenosine monophosphate; G6P, glucose 6-phosphate; GP, glycogen phosphorylase; GS, glycogen synthase; HK, hexokinase; PFK, phosphofructokinase.
Balance equations, rate equations, and parameter estimates of the complete model can be found in our previous theoretical account (DiNuzzo et al, 2010). Note that hexokinase- and phosphofructokinase-catalyzed reactions were lumped together in our previous model. The maximum rates of these enzymes have been obtained by assuming a glucose 6-phosphate steady-state concentration of 0.1 μmol/L in both neurons and astrocytes, whereas rates of glycogen synthase- and phosphorylase-catalyzed reactions have been adjusted for experimental glycogen turnover and detection limit (see text). In rate equations, × is either n (neuronal) or a (astrocytic).
The synthesis and degradation of glycogen are regulated through allosteric inhibitors and activators as well as via reversible phosphorylation–dephosphorylation cascades of glycogen synthase (GS) and glycogen phosphorylase (GP). The changes in phosphorylation status of both enzymes are under hormonal as well as energetic control (Roach, 2002). However, as we were interested in the stimulation-induced changes of glycogen metabolism, we considered stationary hormonal conditions. Moreover, we conformed to prior modeling works (Lambeth and Kushmerick, 2002; Dash et al, 2007) by incorporating the phosphorylation-induced modulation of enzyme activity in the regulatory effects of allosteric ligands. Specifically, adenosine monophosphate (AMP) has the predominant role in the allosteric activation of brain GP (Lowry et al, 1967; Dash et al, 2007; Walcott and Lehman, 2007). Accordingly, brain GP isozyme is more sensitive to control through changes in AMP concentration than through phosphorylation cascade system (Crerar et al, 1995; Pfeiffer-Guglielmi et al, 2003). As even small changes in cellular ATP level associated with increased energy demand result in large variation of the AMP level, the AMP-dependent mobilization of glycogen deposits is exquisitely sensitive to the cell energy status (see Roach, 2002). Because of the major role of AMP in GP control, we neglected the inhibition of the enzyme by several allosteric effectors including glucose, glucose-6-phosphate, ATP, purines, and AMP at nonphysiological concentrations (above 2 μmol/L) (Walcott and Lehman, 2007). Glycogen is mobilized as glucose 1-phosphate, which is in equilibrium with glucose-6-phosphate through a reaction catalyzed by phosphoglucomutase. Conversion of glucose 1-phosphate to glycogen requires the equivalent of one ATP due to the formation of uridine diphosphate glucose by uridine diphosphate-glucose pyrophosphorylase. We included these latter reactions into the GS-catalyzed synthesis of glycogen. In particular, the incorporation of glucose into glycogen can be greatly simplified by considering the tight inverse coupling between GS activity and tissue glycogen content, as clearly demonstrated in the skeletal muscle (Nielsen and Richter, 2003, and references therein), which is coherent with the fact the glycogenesis is a saturable process (Champe and Harvey, 1994). Given the similarity between muscle- and brain-type GS enzyme isoforms, we assumed that changes in brain glycogen content during activation have a primary role in the regulation of GS via a hyperbolic-type relationship (Nielsen and Richter, 2003). This simple dependence circumvents the incorporation of the overall metabolic network for glycogen synthesis.
The connection between astrocytic glycogen metabolism and our previous biochemical model was formalized at the level of glucose-6-phosphate. To model glucose-6-phosphate concentration explicitly, which we assumed to have steady-state value of 0.1 mmol/L (Watanabe and Passonneau, 1973), we linked glycogen metabolism to glycolysis through the separation of the reactions catalyzed by hexokinase (HK) and phosphofructokinase (see Table 1). Rate equations and kinetic parameter estimates for these enzymes were obtained after analysis of metabolite concentration transients as well as resting and activation-induced glucose and lactate flow rates in absence of glycogenolysis. Specifically, we correctly reproduced our original simulation outcomes for glucose and lactate fluxes (DiNuzzo et al, 2010) with the present model—i.e., when glycogen metabolism is inactive. The resulting HK product-inhibition constant (KI,G6P = 0.017 mmol/L) is consistent with the observation that glucose-6-phosphate severely inhibits the enzyme at micromolar concentrations (Liu et al, 1999, and references therein), and is in good agreement with early reports from the mouse brain (Lowry and Passonneau, 1964).
Glycogen phosphorylase is active only when high-energy phosphates are depleted (Lowry et al, 1967), whereas under resting conditions it is >85% inactive (Breckenridge and Norman, 1965). Furthermore, measured flux of glucose through GS is very small (< 3 nmol/g per min) in awake resting conditions (Oz et al, 2003). Therefore, we considered exiguous basal activity of both GS and GP under steady-state resting conditions. As experimental data for maximal reaction rates of these enzymes
We additionally tested the model response after altering the relative neuronal versus astrocytic activation fraction from 1.5:1 to 3:1, matching the same conditions adopted in our previous modeling work (DiNuzzo et al, 2010). Cell activation was shown to have the largest modulatory effect on cellular substrate utilization, with a 1.5:1 ratio colliding with the limitation of astrocytic glucose transport capacity, thus suggesting a biochemical/transport basis for glycogen to be located in these cells (DiNuzzo et al, 2010). Although the 3:1 ratio was identified as a low-end estimate for a sustained neuronal versus astrocytic stimulation based on current literature (DiNuzzo et al, 2010), it is worth exploring higher levels of astrocytic activation as they might hold at a subcellular level or during rapid transients of the energetic demand likely occurring during the early phase of stimulation. Moreover, mobilization of glycogen in astrocytes might circumvent the failure of astrocytic glucose uptake, thus further rationalizing a potentially lower neuronal versus astrocytic stimulation ratio. To examine the intraparenchymal metabolite trafficking, we adopted the convention of expressing cell outward glucose and lactate fluxes as positive and cell inward fluxes as negative. Model simulations were performed by Rosenbrock-based integration algorithms using the software MATLAB (The Mathworks Inc., Natick, MA, USA; http://www.mathworks.com/) version 7.0.4 R14.
Results
First, we examined the activity of the enzymes involved in glycogen metabolism under conditions of stimulation using 1.5:1 neuronal/astrocytic activation ratio, taking into account the rate of brain glycogen synthesis and degradation observed experimentally (Dienel and Cruz, 2006; Oz et al, 2007; Swanson et al, 1992). Under these conditions, glycogenolysis is robustly activated during the stimulation period. Figure 1 shows the simulated velocity of GS- and GP-catalyzed reactions during a sustained 360-second activation, as well as the astrocytic glycogen level change. In agreement with previous findings, our data show that mobilization of the stored sugar is rapidly activated at the stimulation onset, whereas glycogen resynthesis is slower and protracted compared with the duration of the stimulus, which is also consistent with previous reports indicating that potential GP activity in brain far exceeds that of synthase (Dienel et al, 2002, 2007). Tissue glycogen concentration (inset in Figure 1) was found to decrease by ∼0.4 mmol/L (or 12%), thus resulting in a mean breakdown rate of 1.1 μmol/L per s (∼0.07 μmol/g per min), which is compatible with the experimental values ranging from 0.04 to 0.5 μmol/g per min obtained in animal studies and in cell cultures (Dienel and Cruz, 2006; Swanson et al, 1992). Furthermore, the simulated rate of functional glycogen utilization is almost within the experimental detection limit (20% in individual subjects) of the sole study, which examined the role of human brain glycogen during stimulation by 13C nuclear magnetic resonance spectroscopy (Oz et al, 2007). Our simulations showed that, although at rest simultaneous activation of GS and phosphorylase is prevented, brain activation promotes both glycogenolysis and glycogen synthesis at the same time (Dienel et al, 2007; Shulman et al, 2001; Walls et al, 2009). Accordingly, the analysis of net mean breakdown rate as a function of stimulation time suggests that prolonged stimulations are detrimental rather than advantageous to detect the functional changes in glycogen concentration induced by physiological activation (Figure 2). It is noted that Oz et al (2007) found no significant change in the 13C-labeled C1 glycogen signal measured before and after a 20-minute visual stimulation. However, turnover of glycogen outer layers induced ∼30% clearance of the label before stimulation (Figure 3 in Oz et al, 2007), which started several hours after cessation of glucose infusion. This is to be compared with the amount of glucosyl residues readily available to the phosphorylase in the glycogen outer tier, which is ∼35% (Melendez-Hevia et al, 1993). Therefore, the discrepancy between the study of Oz et al (2007) and our findings may be partly explained by the fact that labeled glucosyl residues were retained in the inner, less accessible tiers of the glycogen granules. The simulated net glycogen utilization after 20 minutes of nearly 0.67 μmol/g (inset in Figure 2) results in a 19% signal change, which is near or below the experimental detection limit if we assume concomitant synthesis and degradation within the same glycogen deposits.
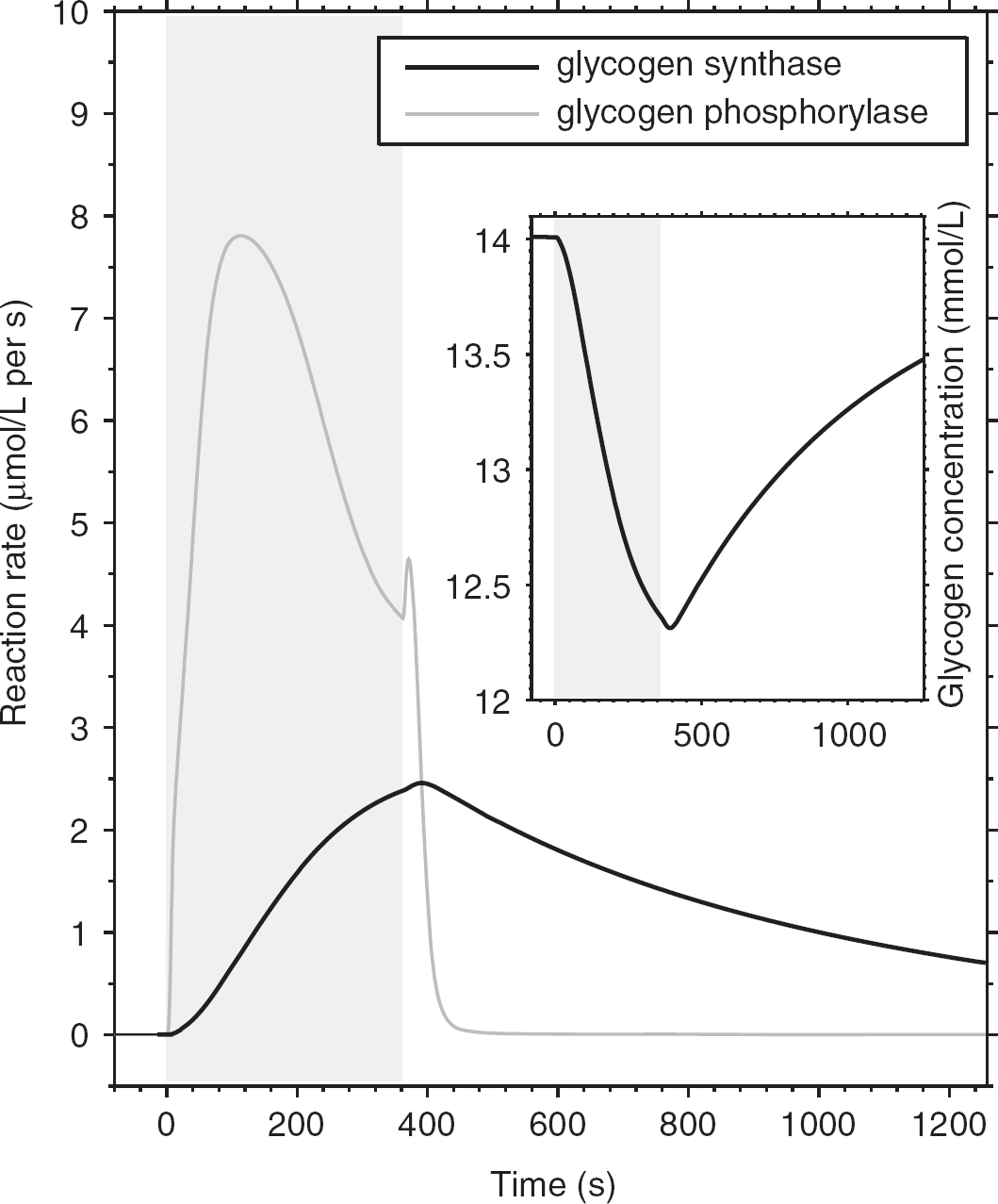
Time courses of glycogen synthase (GS) and glycogen phosphorylase (GP) reaction rates. Brain stimulation produces a rapid activation of GP because of increased energy demand, which effect glucosyl equivalents concentration in astrocytes (inset). The GP-catalyzed mobilization of glycogen decreases during the late phase of the stimulation period, and readily returns to basal level after the end of stimulation. The GS-catalyzed incorporation of glucose into glycogen is delayed and much slower than glycogen breakdown; however, it remains significantly elevated during the poststimulus period. Note that, although the resting activity of both GS and GP is very small, substantial synthesis and degradation of glycogen occur simultaneously during activation. The simulated neuron/astrocyte activation ratio is 1.5:1.
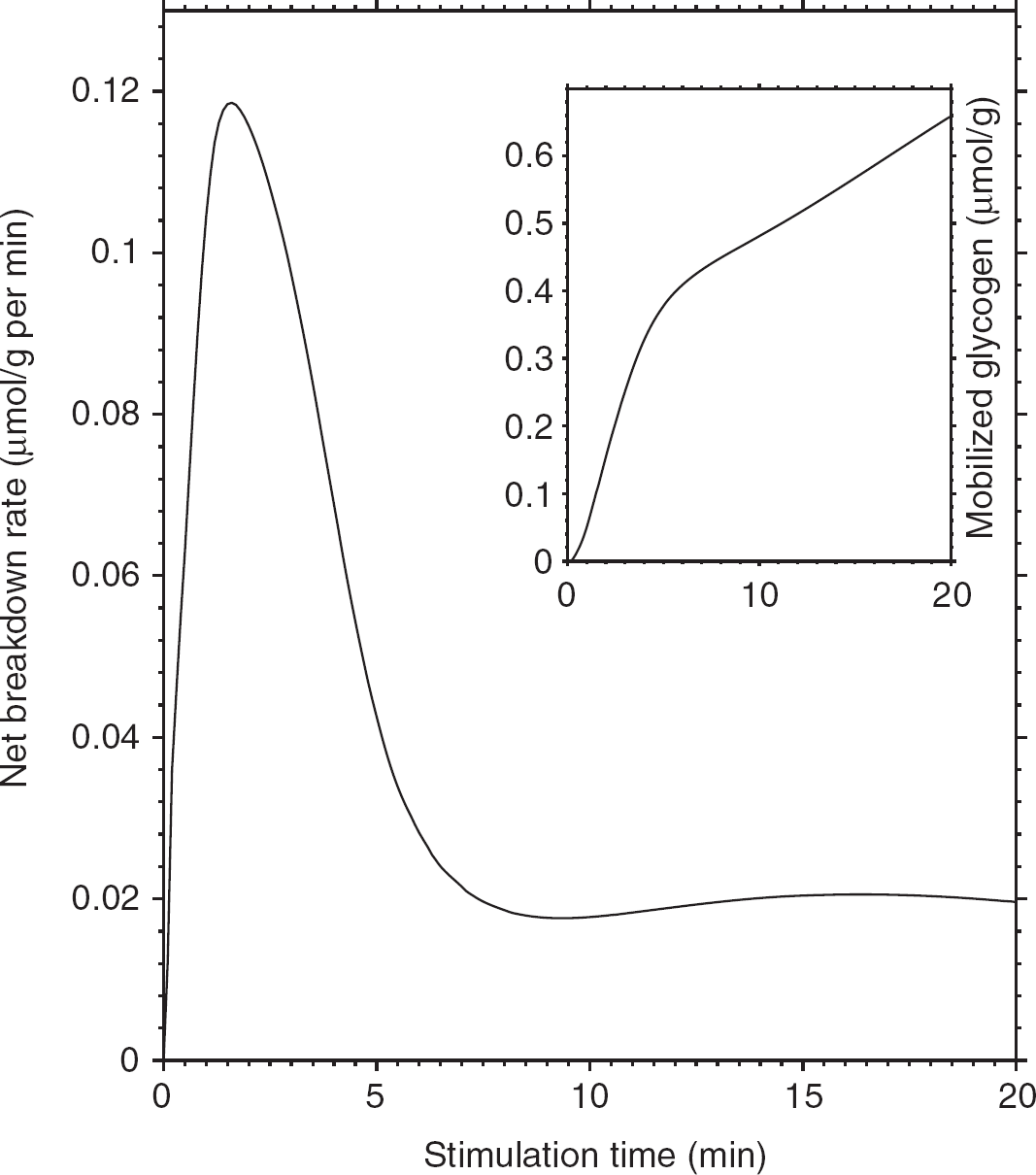
Net brain glycogen breakdown as a function of time during the stimulation. The simulated net glycogen breakdown rate (difference between phosphorolysis rate and synthesis rate) shows a transient peak (up to 0.12 μmol/g per min) at 1 to 2 minutes and then reaches a steady-state value of 0.02 μmol/g per min after 6 minutes. Therefore, the mean net glycogen breakdown rate averaged over the stimulation period decreases with increasing duration of the stimulus. As shown in the inset, the amount of mobilized glycogen is 0.67 μmol/g in correspondence of a 20-minute stimulation. The simulated neuron/ astrocyte activation ratio is 1.5:1.
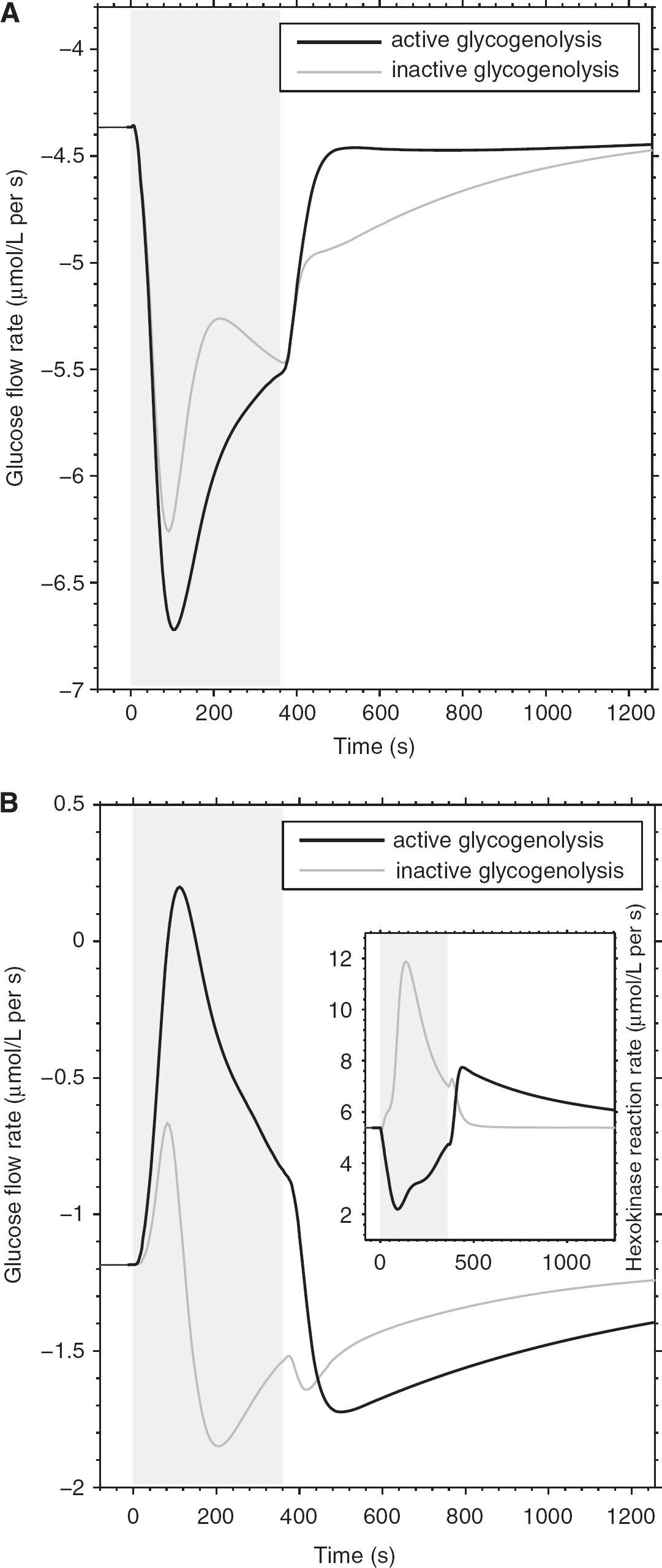
Time courses of neuron (
Figure 3 shows the time course of neuronal and astrocytic glucose flow rates at rest and during brain activation in correspondence of the presence or absence of glycogenolyis. The assumed energetic demand in astrocytes after stimulation was previously shown to rapidly deplete astrocytic glucose due to unbalanced supply and demand of glucose, suggesting a role for glycogen in supporting high activation of these cells (DiNuzzo et al, 2010). Accordingly, we found that the limited glucose transport capacity of astrocytes is circumvented when astrocytic energy production is partly derived from glycogenolysis, as glucose depletion in astrocytes is prevented by glycogen mobilization (data not shown). Nevertheless, the stimulation-induced glycogen breakdown reinforces the simulated glucose flux trends obtained in the absence of glycogen. Specifically, neuronal glucose uptake during activation is further increased by glycogenolysis (Figure 3A), whereas the transport of glucose in astrocytes drops (Figure 3B). Analysis of enzyme activity reveals that phosphorolysis of astrocytic glycogen by GP facilitates the rapid formation of glucose-6-phosphate compared with uptake and phosphorylation of glucose, producing on average an ∼40% substrate flow inhibition through the HK step during stimulation (65% peak value) as compared with resting conditions (inset in Figure 3B). Given an 85% resting HK product inhibition (calculated from Table 1), this translates to a total HK inhibition of ∼91% on average during stimulation (95% peak value), which is in agreement with previous reports for the level of HK product inhibition by glucose-6-phosphate (Lowry and Passonneau, 1964). The suppression of HK activity in turn reduces glucose utilization in astrocytes, and hence the glucose uptake by these cells is depressed due to the passive nature of glucose carrier proteins. These results are consistent with the observed supercompensatory increase in the utilization of blood glucose during activation but not rest when GP is inhibited (Dienel et al, 2007) (see also Table 2). The astrocytic glucose flow rate shows a small, transient glucose release during the early phase of stimulation, indicating that glucose phosphorylation inhibition is sufficient for astrocytes to export part of their own glucose pool. In the late phase of stimulation and after the end of stimulation, glucose is directed to glycogen first rather than directly entering glycolysis, thus bringing about increased astrocytic and decreased neuronal glucose uptake compared with inactive glycogenolysis.
Mean substrate flow rate changes as a function of cell stimulation and glycogen metabolism
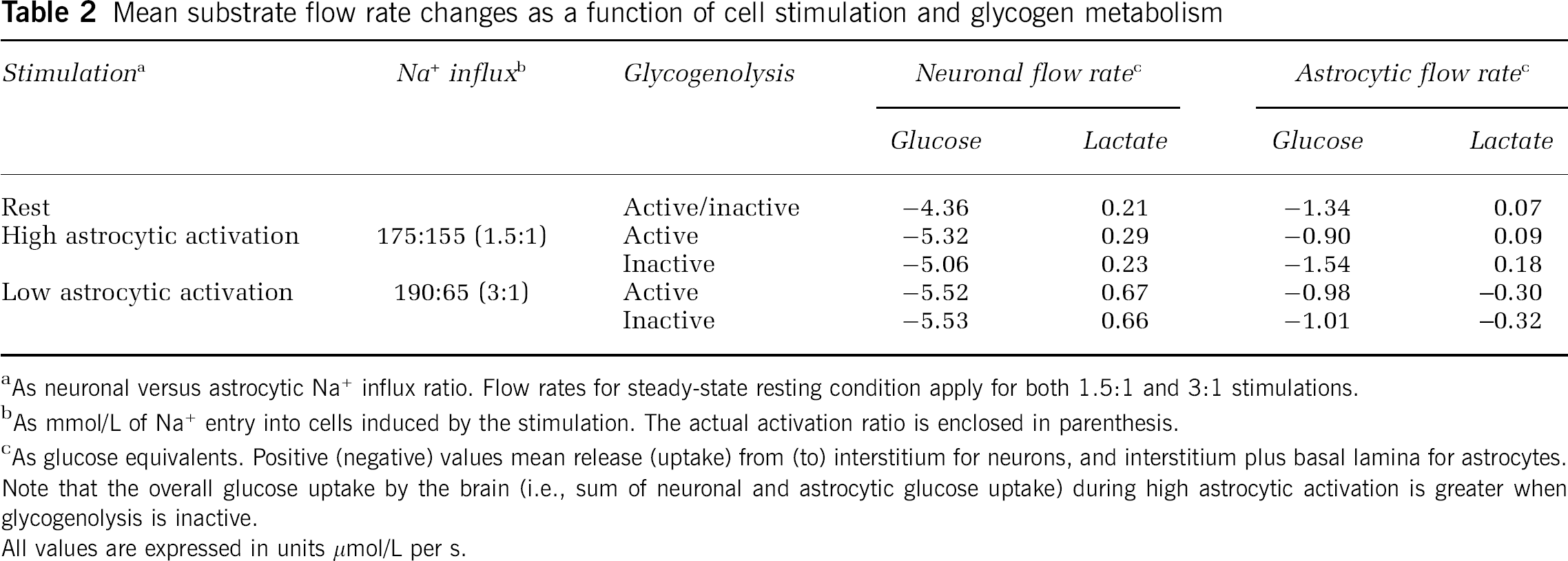
As neuronal versus astrocytic Na+ influx ratio. Flow rates for steady-state resting condition apply for both 1.5:1 and 3:1 stimulations.
As mmol/L of Na+ entry into cells induced by the stimulation. The actual activation ratio is enclosed in parenthesis.
As glucose equivalents. Positive (negative) values mean release (uptake) from (to) interstitium for neurons, and interstitium plus basal lamina for astrocytes.
Note that the overall glucose uptake by the brain (i.e., sum of neuronal and astrocytic glucose uptake) during high astrocytic activation is greater when glycogenolysis is inactive.
All values are expressed in units μmol/L per s.
Next, we examined the contribution of glycogen mobilization in astrocytes for the provision of energy substrate for neurons in the form of lactate. Figure 4 shows the time course of the resting and stimulation-induced neuronal and astrocytic lactate flow rates in correspondence of the presence or absence of glycogenolysis. Simulations show negligible differences in intercellular lactate flux induced by the metabolism of glycogen, especially at the onset of increased activity (prevalence of glycogenolysis) relative to the late and poststimulation periods (prevalence of glycogenesis), both in neurons (Figure 4A) and astrocytes (Figure 4B). This indicates that glycogen-derived pyruvate is consumed by astrocytes rather than transferred to neurons as lactate, supporting the hypothesis that shuttled lactate might have a minor role as a cell-to-cell energy substrate (DiNuzzo et al, 2010). In addition, we found that the rise in lactate concentration induced by stimulation is independent of glycogen breakdown (data not shown), consistent with experimental findings (Caesar et al, 2008). This further suggests that lactate accumulation is governed essentially by energy demand and that substrate availability is unrestricted for both neurons and astrocytes.
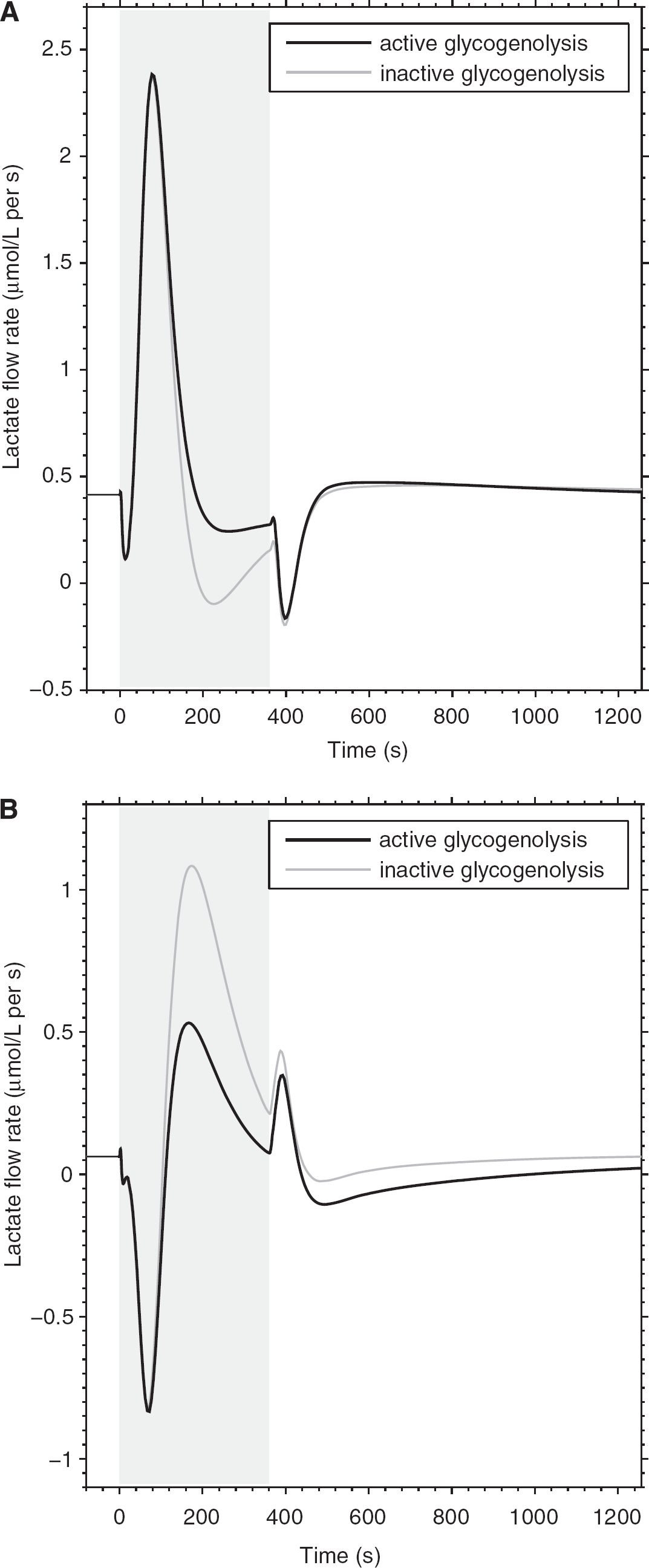
Time courses of neuron (
Finally, to analyze the role of glycogenolysis in astrocytes during different levels activation of these cells, we calculated the mean glucose and lactate flux as a function of the neuronal versus astrocytic stimulation (Table 2). A neuronal versus astrocytic stimulation-induced Na+ influx ratio of 190:65 mmol/L (i.e., condition of ‘low’ astrocytic activation, or 3:1 neuronal/astrocytic activation ratio) was found to result in glycogen-insensitive substrate utilization for both neurons and astrocytes, which reflects a smaller increase of intracellular AMP concentration after astrocytic energy consumption and a consequently reduced glycogen breakdown. On the contrary, for a neuronal versus astrocytic stimulation-induced Na+ influx ratio of 175:115 mmol/L (i.e, condition of ‘high’ astrocytic activation, or 1.5:1 neuronal/astrocytic activation ratio), glycogen mobilization induces a mean 0.64 μmol/L per s decrease in astrocytic glucose uptake, which accompanies a 0.26 μmol/L per s increase in glucose routing to neurons. Simulations showed also the flux of intermediary metabolite as lactate depends on the level of stimulation of neurons compared with astrocytes. In particular, under low activation conditions of astrocytes, neuronal lactate release is 2.5-fold higher relative to conditions of high astrocytic stimulation. Accordingly, astrocytic glycogen-derived carbons are partly replaced by lactate taken up (flow rate of 0.30 μmol/L per s) from the extracellular space—that is, shuttled from neurons to astrocytes—which is in agreement with previous theoretical reports (Simpson et al, 2007; Mangia et al, 2009b). In this condition, glucose equivalents as transferred lactate represents ∼30% of astrocytic glucose uptake, whereas high activation of astrocytes compared with neurons results in lactate release by both cell types. Taken together, these simulations indicate that glucose is preserved for neuronal utilization regardless of the engagement of astrocytes, with lactate acting as a versatile (albeit secondary) substrate for either utilization (i.e., oxidation) or disposal (washout by the bloodstream) (DiNuzzo et al, 2010).
Discussion
On the basis of the results of this study, we propose a novel contribution to the functions of brain glycogen under physiological conditions. First, as generally accepted, glycogen buffers the provision of glucose-6-phosphate to provide energy substrate for astrocytic glycolysis, and possibly precursor for pentose phosphate pathway (see below). Second, increased glucose-6-phosphate level would reduce the astrocytic use of glucose delivered by blood during glycogenolysis due to inhibition of HK, thus allowing neurons greater access to glucose provided by blood. Third, some of the unmetabolized intracellular glucose in astrocytes could now diffuse to neurons as supplementary fuel.
Therefore, the proposed mechanism for the suppression of astrocytic glucose consumption identifies glycogen as a modulator of the changes in cellular glucose availability during stimulation. Specifically, reduction in the amount of glucose taken up and metabolized by astrocytes is realized via the inhibitory feedback on HK exerted by glucose-6-phosphate, which increases substantially as a result of stimulation-induced glycogenolysis (Figure 1). Noteworthy, even the mobilization of 0.4 μmol/L glucosyl residues is found to produce an ∼40% HK flux inhibition (relative to rest, averaged over the entire stimulation period) in astrocytes, with a consequent routing of glucose to neurons (Figure 3). Interestingly enough, intercellularly shuttled lactate is found to be fairly independent on glycogen utilization (Figure 4; Table 2), indicating that brain metabolism is efficient in relocating blood-borne glucose to satisfy cellular energy demand, with only secondary dependence on carbon equivalents derived from transferred lactate. Simulations showed that glycogen mobilization can induce partial release of the astrocytic glucose pool (Figure 3B), which is contrary to the notion that astrocytes cannot release glucose because glucose-6-phosphatase activity in the brain is very low (Brown, 2004). Accordingly, glucose-6-phosphatase is not included in the present model, as no significant dephosphorylation of glucose-6-phosphate is found in the brain
Our results suggest that glycogen acts as a buffer within astrocytic metabolism to avoid the competition for glucose between neurons and astrocytes, thereby reducing the impact on neuronal metabolism by varying astrocytic energy demand. Accordingly, the assumption about cell stimulation ratio becomes much less critical on model outcomes (Table 2, compare with the results obtained in DiNuzzo et al, 2010), which suggests a role for glycogen in reducing the sensitivity of the brain metabolic response on the potential cellular heterogeneity. It should be realized that, although the simulated increase in glucose availability to neurons after glycogenolysis in astrocytes is relatively modest (from Table 2 it is calculated as a 0.2 mmol/L decrease and 0.1 mmol/L increase of astrocytic and neuronal glucose uptake, respectively, for the entire stimulation interval), the effect can become important
The outcomes of the present theoretical study support the notion that during the early phase after brain stimulation, the properties and the regulations of cellular metabolic and transport competence favor the channeling of blood-borne glucose, rather than glycogen-derived lactate to activated neurons. Notably, the assumption that astrocytes release glycogen-derived lactate (Brown, 2004; Pellerin et al, 2007) is based on findings obtained in cultured cells often exposed to extreme stimulation paradigms or nonphysiological conditions (low or zero glucose concentration) (Brown and Ransom, 2007; Dringen et al, 1993), which might upregulate the reduction of glycogen-derived pyruvate to lactate compared with oxidation in the tricarboxylic acid cycle. Therefore, the correlation between the rate of lactate release and glycogen breakdown observed in these studies with possibly altered metabolic demand is not in contradiction with our modeling conclusions.
Although there is no thermodynamic energetic benefit for astrocytes to mobilize glycogen when glucose is available as a substrate, mobilization of glycogen has the clear kinetic advantage of rapidly providing energy for the fast energetic needs of astrocytes, such as K+ sequestration after neuronal action potentials. In fact, K+ was found to robustly stimulate astrocytic glycogenolysis (Dienel and Cruz, 2006, and references therein). Moreover, glycogen may sustain the net synthesis of glutamine from glycogen (Gibbs et al, 2008) via stimulation of the anaplerotic pyruvate carboxylation pathway in astrocytes, as well as the generation in the pentose phosphate pathway of the NADPH needed for the detoxification of reactive oxygen species (Murin et al, 2009, and references therein). The latter point applies both to neurons, which can divert a larger fraction of glucose to pentose phosphate pathway during activation, and to astrocytes, as involvement of glycogen for disposal of peroxides was directly demonstrated in astrocytic preparations (Rahman et al, 2000). On the other hand, our results suggest that part of the functional significance of brain glycogen is identified in the energetic benefit for neurons (i.e., supporting neuronal glycolysis to proceed). This is perhaps necessary to maintain neuronal processes depending on transmembrane sodium gradient, which are thought to use preferentially glycolytic energy (Ames, 2000). Accordingly, glucose is essential for the maintenance of neural activity in cultured neurons (Bak et al, 2009) and in rapidly prepared brain slices regardless of the ATP concentration (Yamane et al, 2000). Notably, the different dependence of brain glycogen utilization on the degree of astrocytic activation adds another level of complexity to the composite character of the metabolic response of the brain to stimulation. Although definite experimental proof about the physiologic significance of preserving extracellular glucose for neuronal use remains elusive, we conclude that the theoretical findings of this study support the importance of glucose as neuronal energy substrate during increased neuronal activity.
Footnotes
Acknowledgements
The authors thank Gulin Oz for her helpful comments on the paper.
The authors declare no conflict of interest.