
Abstract
To assess the role of tumor necrosis factor-α (TNF-α) in modulating the process of cerebral ischemic injury, we identified TNF-α-producing cells and studied the time course of TNF-α expression. Immunoreactivity for TNF-α appeared in white matter of the mouse hippocampus as early as 1.5 h following a 30-min global ischemic insult. Double staining for TNF-α and glial fibrillary acidic protein (GFAP) suggested that the TNF-α-positive cells are most likely microglia, not astrocytes. TNF-α immunostaining decreased at 6 and 24 h but increased again at 3 days, when pyramidal neurons showed degeneration. Adjacent-section staining for microglia and double staining with GFAP suggested that TNF-α-positive cells in the pyramidal cell layer were microglia and those in the white matter were astrocytes. By 5 days TNF-α immunostaining disappeared from these glial cells, while a number of microglia were accumulated in the degenerated hippocampal pyramidal layer. Pyramidal neurons never expressed TNF-α immunoreactivity. Western blotting confirmed biphasic TNF-α expression. Our findings suggest that early production of TNF-α by microglia may activate a cytokine network in postischemic brain resulting in TNF-α synthesis by astrocytes.
Several pathophysiologic factors contribute to ischemic brain injury, such as excitotoxicity, intracellular calcium overload, altered gene expression, and reactive oxygen species (Matsuyama et al., 1993). Recently, cytokines also have become a focus of attention (Minami et al., 1992; Wiessner et al., 1993; Liu et al., 1994). Tumor necrosis factor-α (TNF-α) is a pleiotropic cytokine with diverse functions mediating the inflammatory and immune responses in injury and repair. TNF-α has been linked to oligodendroglial injury, to cytotoxicity of lipopolysaccharide-stimulated astrocytes, and to astrocytic proliferation in vitro (Robbins et al., 1987; Selmaj and Raine, 1988). It enhances leukocyte adherence to endothelium, probably through activation of adhesion molecules such as intercellular adhesion molecule-1 (Warren 1990; Mantovani et al., 1992). TNF-α activates procoagulant processes (Pober and Cotran, 1990) and stimulates macrophages to produce other immune mediators (Hori et al., 1989).
TNF-α, produced mainly by activated macrophages, also is produced by microglia and astrocytes in culture (Lieberman et al., 1989; Sawada et al., 1989; Chung and Benveniste, 1990). In addition, evidence has been accumulated that glial cells contribute to a cytokine network in the central nervous system (Benveniste, 1992; Feuerstein et al., 1994; Rothwell and Hopkins, 1995). Although cerebral ischemic injury promotes glial proliferation, its effects on the production of cytokines have not been fully elucidated. In addition, the glial source of TNF-α has not been identified in vivo, although TNF-α induction in neurons has been reported after focal cerebral ischemia (Liu et al., 1994).
Production of cytokines including TNF-α has been investigated in traumatic brain injury models (Taupin et al., 1993; Shohami et al 1994), in which such cytokines are rapidly synthesized in the brain as early as 3 h following the insult. In cultured astrocytes, a hypoxia/reoxygenation insult induces synthesis of cytokines as early as 1.5 h (Maeda et al., 1994; Matsuo et al., 1995). The in vivo time course of cytokine synthesis has not been investigated in the ischemia/reperfusion model.
This study was sought to immunocytochemically determine the time course and the cellular sources of TNF-α production in the brain following transient global ischemia.
MATERIALS AND METHODS
Animal preparation
A total of 266 male Balb/c mice (25–30 g; Charles River, Yokohama, Japan) underwent the cerebral ischemic insult in this study. One hundred animals were used to examine the relationship between their neurologic status and the hippocampal injury following cerebral ischemia. The analysis for brain TNF-α immunoreactivity following ischemia was performed with the remaining 166 animals.
Mice were anesthetized lightly by ether inhalation, and cerebral ischemia was produced by occluding both common carotid arteries with microaneurysm clips for 30 min. Rectal temperatures were maintained at 37.0–37.2°C during the insult and for at least 1 h following the ischemia. The neurologic status of each animal was recorded during the 30-min period of occlusion. Disturbances of consciousness, piloerection, ptosis, defective eye closure, jumping, running, rolling, and splayed-out hindlimbs variously were observed. Animals were divided into two groups: (1) comatose (showing disappearance of the righting reflex) and (2) noncomatose. In some animals CBF was measured by a laser-Doppler apparatus (Neuroscience, Japan) in a middle cerebral artery distribution bilaterally during and for 1 h following carotid occlusion.
Immunocytochemistry
Animals were perfusion-fixed with periodate lysine-paraformaldehyde solution under pentobarbital anesthesia (50 mg/kg body wt) at 1.5 h (n = 5), 6 h (n = 4), 24 h (n = 5), 3 days (n = 5), and 5 days (n = 5) following clip removal. Sham-operated animals (n = 5) whose carotid arteries were exposed but not clipped were perfused with the same fixative. The brain was removed and sectioned at 20 μm on a vibratome. Sections were incubated overnight at 4°C with a 1:500 dilution of monoclonal anti-mouse TNF-α antibody (Upstate Biotechnology, NY, U.S.A.; lot 12391). They were then stained by the avidin-biotin-peroxidase complex method (Vectastain ABC Kit, Vector, NY, U.S.A.). TNF-α immunoreactivity was identified with a diaminobenzidine reaction. To test for specificity, Western blotting and omission and absorption control experiments were performed. Absorption was carried out using a mixture of primary antibody and recombinant mouse TNF-α (10–6 M; Genzyme, Cambridge, MA, U.S.A.).
Adjacent sections were incubated with a monoclonal antibody against a macrophage (F4/80, 1:10; Serotec Ltd, U.K.) to detect microglia or were incubated with an anti-GFAP antibody (1:500; Dako, Denmark) to identify astrocytes. Some single sections were doubly stained for TNF-α and GFAP. In this case, GFAP immunoreactivity was detected using fluorescein isothiocyanate–conjugated goat anti-rabbit IgG (Vector, Burlingame, CA, U.S.A.) as a second antibody after TNF-α staining and was observed under a fluorescence microscope.
Western analysis
Animals were decapitated after sham operation and at 1.5 h, 24 h, and 3 days following ischemia (5 animals/group), and the hippocampi were removed. Tissue was homogenized with sample buffer containing 2% sodium dodecyl sulfate. Samples containing 9 μg of protein were subjected to 10% polyacrylamide gel electrophoresis in the presence of 0.1% sodium dodecyl sulfate. Protein bands were transferred from the gel to a nitrocellulose membrane using a semidry transfer apparatus and a blotting buffer including 0.192 M glycine and 20% methanol (pH 8.4). The membrane then was incubated with a 1:500 dilution of primary antiserum and stained by the avidin-biotin-peroxidase complex method as described. Protein determinations were performed in triplicate with a colorimetric dye-binding assay (Bio-Rad, Richmond, CA, U.S.A.). The quantitative analysis for TNF-α immunoreactivity was carried out using a computerized densitometry system (ATTO Densitograph, ver. 4.0, Japan).
Statistical analysis
TNF-α-immunoreactive cells were counted on 12 sections including hippocampus per animal (five animals in groups at 1.5 h, 24 h, 3 days, and 5 days and four in group at 6 h). Results were expressed as cells per square millimeter ± SD. Statistical analysis of the data was done by an analysis of variance with a post hoc Scheffé test.
RESULTS
Neurologic status
The correlation between neurologic status of mice during ischemia and histologic appearance after clip removal is shown in Tables 1 and 2. Fifty-three of 100 animals showed loss of the righting reflex during ischemia. Twenty-seven animals of these 53 died during ischemia. Twenty-four of the 26 surviving mice showed hippocampal neuronal degeneration 4 days after ischemia, while 2 did not. Thus, only 24% of animals undergoing carotid occlusion could be used in immunocytochemical experiments.
Neurologic assessment during ischemia

Histologic assessment after ischemia

Monitoring CBF during ischemia in a separate group showed a high correlation between neurologic status and CBF decrease (Fig. 1). Carotid occlusion resulted in a bilateral CBF decrease by 10–20% in all comatose animals, while noncomatose animals showed a CBF decrease by 40–60% during ischemia.
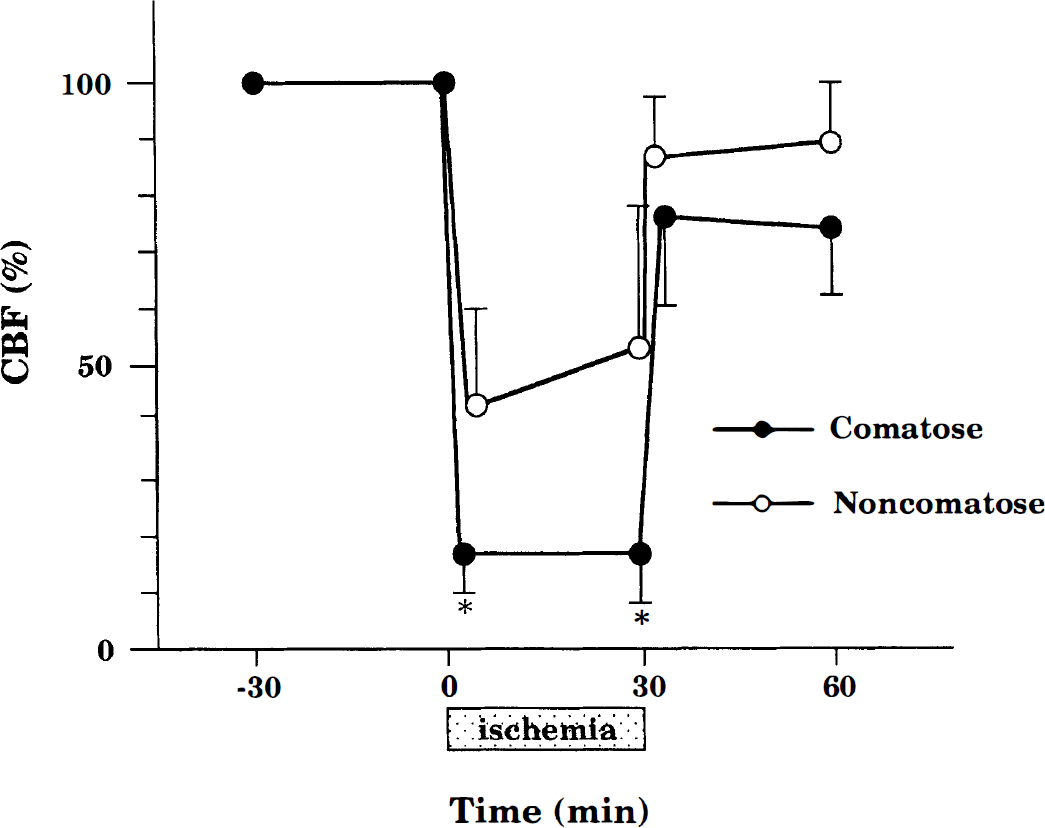
Regional CBF during 30 min of bilateral common carotid occlusion and 30 min of reperfusion in comatose and noncomatose mice. Animals were operated initially under halothane anesthesia. Regional CBF was determined by laser-Doppler flowmetry. The tip of the probe was placed bilaterally on the intact skull over the ischemic cortex (1 mm anterior and 4.5 mm lateral from bregma). The individual data are presented as mean values of right and left CBF, •, comatose mice (n = 6); ○, noncomatose mice (n = 5). There are significant differences between groups (*p < 0.01).
Immunocytochemistry for TNF-α
Of 166 animals subjected to bilateral carotid ligation, 39 showed coma but survived.
In sham-operated animals, almost no TNF-α was expressed in hippocampal cells (Fig. 2A). Following ischemia, TNF-α immunoreactivity was found exclusively in the hippocampal formation including the dentate gyrus. Two appearances characterized the immunoreactive cells, one being a round cell with few processes (Fig. 3 A, B, and C) and the other a pyramidally shaped cell with many processes (Fig. 3E). The former TNF-α-positive cells were scattered sparsely in the hippocampal white matter 1.5 h following ischemia, and the latter were found late in the recirculation period at 3 days. The round cell type was found in the pyramidal cell layer at 1 and 3 days after ischemia (Fig. 3A and B). TNF-α immunoreactivity also was evident in cells adjoining blood vessel lumina, most likely endothelial cells (Fig. 3B), but was never seen in pyramidal neurons or in granule cells of the dentate gyrus. This immunoreactivity was not seen in sections incubated without the primary antibody or with antibody solution absorbed using recombinant mouse TNF-α, confirming the specific expression of TNF-α in glial cells following ischemic insult (Fig. 4).
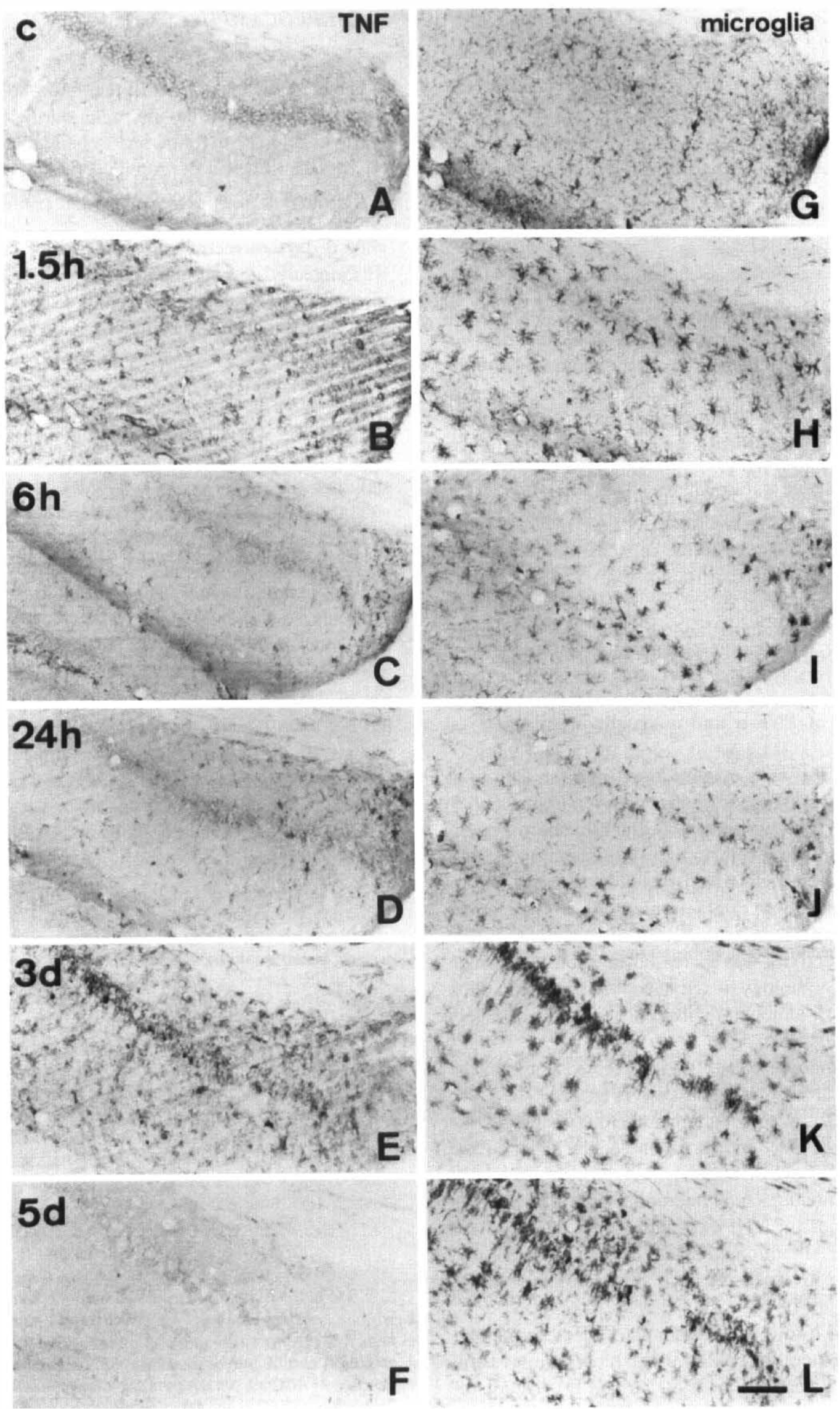
Distribution and time course of immunoreactivity for tumor necrosis factor-α (TNF-α) (
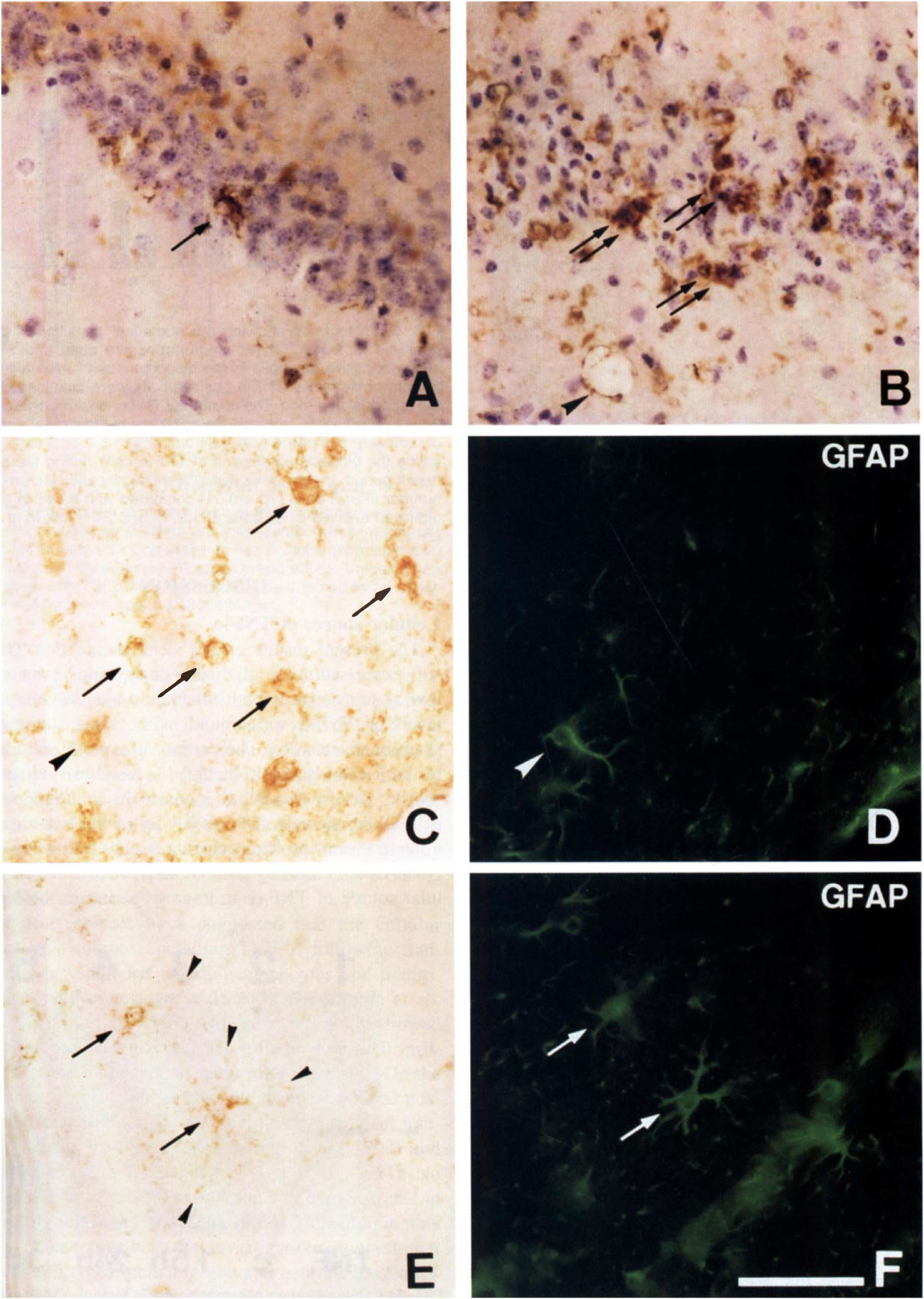
Photomicrographs of postischemic mouse hippocampus immunolabeled with antibody against tumor necrosis factor-α (TNF-α) (
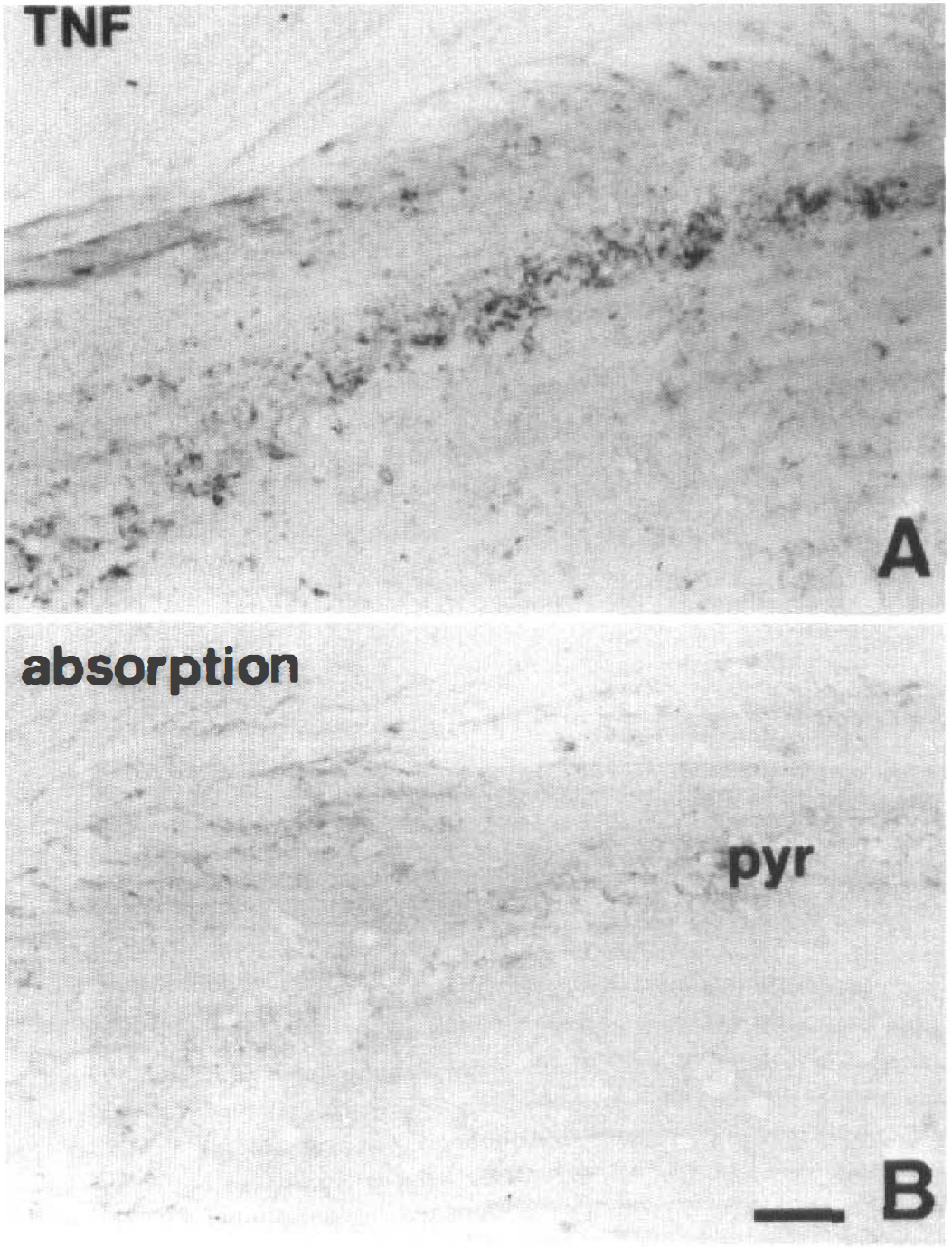
Representative photomicrographs showing the result of absorption. Hippocampal sections obtained from a mouse 3 days following 30 min of ischemia were reacted with tumor necrosis factor-α (TNF-α) antibody (
Double staining for TNF-α and GFAP revealed pyramidally shaped TNF-α-positive cells staining for GFAP at the same time, but also round small TNF-α-positive cells that did not (Fig. 3C–F). This indicates that both GFAP-positive astrocytes and another glial cell type, such as microglia, express TNF-α following ischemia.
Time course of TNF-α and microglia staining
The number of cells staining over time for TNF-α or F4/80 in the dorsal hippocampus appear in Figs. 2 and 5. In sham-operated animals, F4/80 antigens were expressed in many glial cells in the white matter (Fig. 2G). Their morphologic characteristics followed the classic morphology of resting ramified microglia with a number of long, thin processes. After 1.5 h of recirculation, F4/80 immunoreactivity was increased in microglia, which showed the morphology of activated microglia with thick short processes, although their number remained largely unchanged (Fig. 2H).
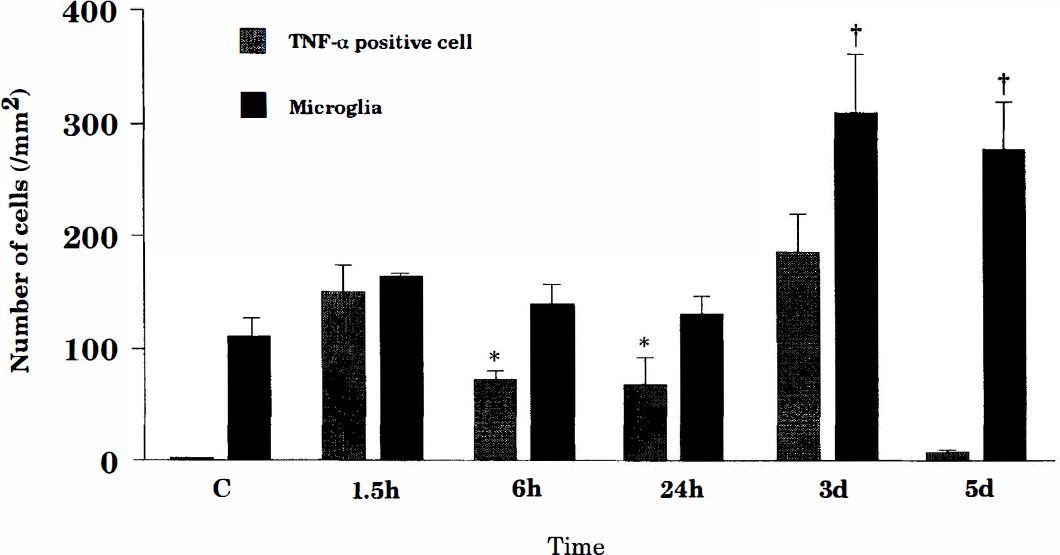
Histograms showing the numerical densities of tumor necrosis factor-α (TNF-α)–immunopositive cells and F4/80-immunopositive microglia in the hippocampus of mice following 30 min of ischemia. TNF-α-positive cells were identified during the reperfusion period. The number of TNF-α-positive cells at 6 or 24 h is significantly reduced (*p < 0.01 by multifactorial analysis of variance) compared with the number at 1.5 h or 3 days, indicating the biphasic expression of TNF-α cells during the reperfusion period. In contrast, microglia are significantly increased in number after 3 days (†p < 0.01) compared with those of sham-operated animals or animals at 1.5, 6, and 24 h following ischemia.
TNF-α immunoreactivity was expressed anew in the hippocampus at 1.5 h of reperfusion (Fig. 2B). The TNF-α-positive cells were predominantly distributed in the white matter of CA1 and in the molecular layer of the dentate gyrus. The distribution pattern was similar to that of F4/80 immunoreactive microglia (Fig. 2B and H). Almost none of the TNF-α-immunopositive cells were labeled by GFAP antibody. These findings indicate that the TNF-α-positive cells at 1.5 h are microglia (Fig. 3C and D).
At 6 and 24 h following ischemia, TNF-α immunoreactivity was generally reduced in the hippocampus compared with that seen earlier (Fig. 2C and D). The distribution pattern also was changed. TNF-α-positive microglia and endothelial cells had decreased significantly in the whole hippocampus (Fig. 5), while the number of TNF-α-positive round cells within the pyramidal layer had slightly increased.
On the other hand, no further morphologic changes were conspicuous in the microglia during the period between 6 and 24 h following ischemia. Some of these microglia still had hypertrophic perikarya and short thick processes and were seen in the pyramidal cell layer (Fig. 2I and J).
At 3 days of recirculation, staining for TNF-α again was increased in both the pyramidal layer and the white matter regions such as the stratum oriens, stratum radiatum, and lacunosum moleculare, especially in the CA1 sector. Some pyramidal neurons had degenerated in the CA1 layer, where many round small cells staining intensely for TNF-α had accumulated (Fig. 2E). In contrast, in the white matter, pyramidally shaped TNF-α positive cells were increased in number and intensity of staining.
The distribution of microglia was very similar to that of TNF-α-positive cells 3 days after the ischemia. Microglia increased in number both in the pyramidal layer and in the white matter. The microglia in the pyramidal layer showed amoeboid morphology (Fig. 2K). However, most of the microglia in the white matter had many thick processes. Double-staining immunocytochemistry revealed that TNF-α-positive cells in the pyramidal layer did not stain for GFAP, while most TNF-α cells in the white matter also stained for GFAP, indicating that these latter cells were astrocytes (Fig. 3E and F).
At 5 days of recirculation, almost no staining for TNF-α was seen in the hippocampus, although the microglia had continued to accumulate in the pyramidal layer, where degeneration of neurons was clearly evident (Fig. 2F and L).
Densities of TNF-α-positive cells and microglia (cells/1 mm2) in the CA1 region of the hippocampus were 2.5 ± 2.9 and 108.3 ± 20.8 in controls, 148.4 ± 25.7 and 162.5 ± 3.5 at 1.5 h, 72.0 ± 8.4 and 152.0 ± 18.2 at 6 h, 66.3 ± 25.6 and 129.0 ± 16.7 at 24 h, 185.0 ± 33.4 and 310.0 ± 51.8 at 3 days, and 16.3 ± 4.3 and 278.0 ± 41.3 at 5 days, respectively.
Western analysis
A characteristic accumulation of immunoreactive TNF-α was evident as a single band of molecular mass 17 kDa (Fig. 6). The signal was transiently increased during 1.5 h and 3 days after ischemia. The densitometric analysis revealed that the signal intensity was significantly stronger at 1.5 h and at 3 days than that of control (p < 0.01), but did not differ at 24 h from that of control (Fig. 7). This expression pattern was in agreement with the biphasic expression of TNF-α positivity in cells of the hippocampus following the ischemic insult.
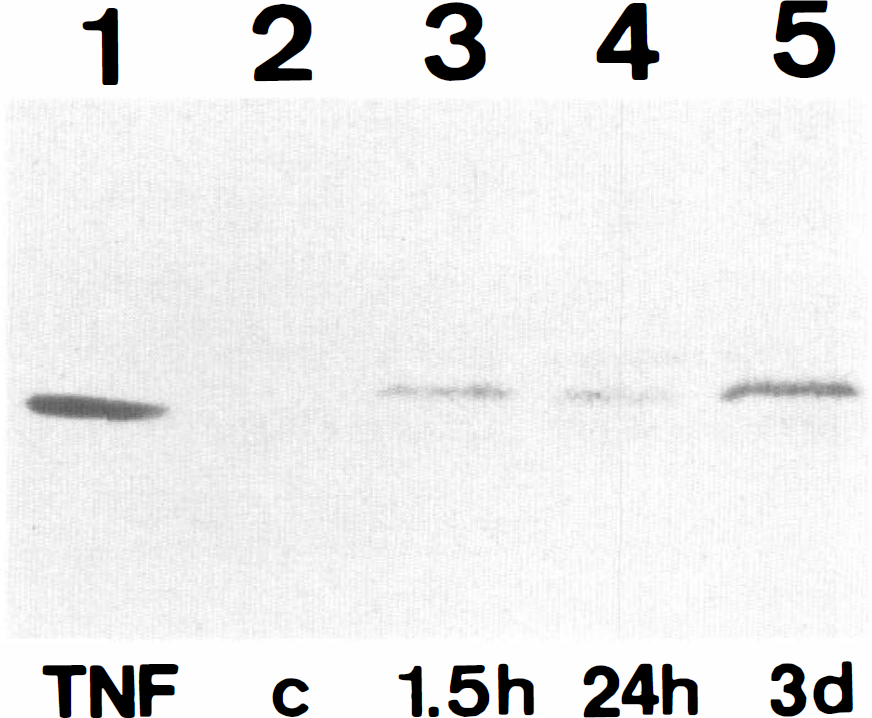
Immunoblotting of tumor necrosis factor-α (TNF-α) from mouse hippocampus. Tissues were prepared from a sham-operated animal (lane 2) and animals at 1.5 h (lane 3), 24 h (lane 4), and 3 days (lane 5) of recirculation. Recombinant mouse TNF-α (20 ng) was included in Lane 1. A specific band for molecular mass of 17 kDa is induced after ischemia. The intensity of staining is reduced at 24 h and increased again at 3 days.
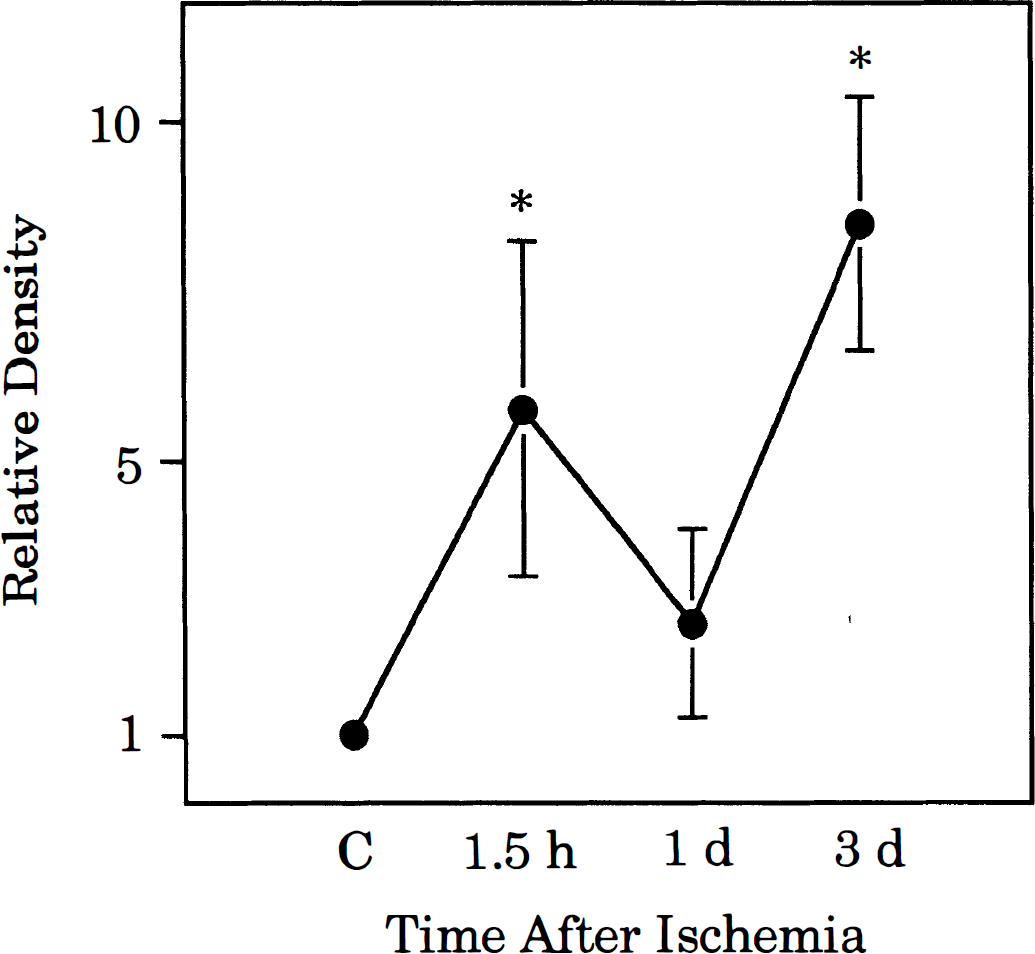
Quantitative evaluation of tumor necrosis factor-α (TNF-α) immunoblotted by Western analysis. Symbols indicate means and SDs for determination made on five animals expressed relative to the density detected in control preparations. Values differing significantly (*p < 0.01) from control, determined by an analysis of variance with post hoc Scheffé test.
DISCUSSION
Cellular source of TNF-α
The present results clearly demonstrate that TNF-α was expressed in the glial cells of the hippocampus following transient cerebral ischemia, while only very low levels of TNF-α were found in the same structure in sham-operated mice. The pattern of expression induced by an ischemic insult included a very early transient profile with a maximum increase in immunopositive cells taking place by the first day following ischemia and a late transient profile occurring 3 days following ischemia. Double staining with GFAP revealed that the cellular source of TNF-α at an early period in the hippocampus was more likely microglia than GFAP-positive astrocytes, and the sources in the late period were both astrocyte and microglia. Previous in vitro studies have shown that TNF-α is synthesized by brain cells such as microglia (Giulian et al., 1986; Frei et al., 1987; Sawada et al., 1989) and astrocytes (Lieberman et al., 1989; Chung and Benveniste, 1990). The present data are in agreement with the results in culture mentioned already and further reveal a biphasic expression of TNF-α during reperfusion by these two cell types.
Previous studies have suggested that the cellular source of cytokines, including TNF-α, following traumatic injury included invading monocytes and neutrophils, as well as resident brain cells (Woodroofe et al., 1991; Taupin et al., 1993). However, this study showed no histologic evidence for such inflammatory cell infiltration in the hippocampus early after the insult. On the other hand, recent studies have indicated neurons as possible cellular sources of TNF-α following surgical traumatic lesions in mice (Tchelingerian et al., 1993) and persistent focal ischemia in rats (Liu et al., 1994). No neuronal TNF-α protein expression was demonstrated in the brain following transient global ischemia in this study. Taken together, the investigations suggest that various cell types including glia and neurons have a capacity to produce TNF-α, depending on specific characteristics of the injury.
Early production of TNF-α
The present study showed a very early production of TNF-α by microglia following the ischemic insult. The intensity of immunoreactivity peaked at 1.5 h and was already declining at 6 h. Similar TNF-α response profiles have been reported in cultured microglia or astrocytes stimulated with lipopolysaccharide (Sawada et al., 1989; Sharif et al., 1993), in traumatized brains (Young et al., 1992; Shohami et al., 1994), and in ischemic brain (Liu et al., 1994). In a model of ischemia/reperfusion injury, Minami et al. (1992) recently have demonstrated an early (30–240 min) induction of mRNA coding for interleukin-1β in cerebral cortex, striatum, and hippocampus following transient forebrain ischemia. The rapid activation of the cytokine gene by ischemia shown in this study and others suggests that immediate early genes such as c-fos and nuclear factor-κB may be important in initiating the cytokine metabolic cascade (Schreck et al., 1991; Kaltschmidt et al., 1994). On the other hand, the rapid decline of brain cytokine levels despite continued presence of glial cells suggests the operation of an endogenous control system producing specific cytokine inhibitors (Dinarello, 1992) and/or other potential antiinflammatory factors such as transforming growth factor-β (Morganti-Kossmann et al., 1992).
Both astrocytes and microglia proliferate in the injured area after an ischemic insult. The mouse model of transient global ischemia used in this study can produce hippocampal neuronal death similar to delayed neuronal death in the rat and gerbil models (Matsuyama et al., 1994a, 1995). In such models, the number of GFAP-positive astrocytes increases throughout the hippocampus. However, this glial response appears relatively late in the recirculation period, some 24–48 h after ischemia. In contrast, microglial activation becomes visible within 30 min of reperfusion according to the lectin reaction (Morioka et al., 1991). It is well known that resting microglia rapidly transform to an activated state in response to a wide range of injuries (Gehrmann et al., 1995). Microglial activation is apparent as proliferation and recruitment to the site of injury, as well as morphologic, immunophenotypic, and functional changes. The last category may include release of mediator substances including cytotoxic compounds such as reactive oxygen intermediates or proteases (Colton and Gilbert, 1987; Thery et al., 1991; Matsuyama et al., 1994b) as well as inflammatory cytokines such as interleukin-1, interferon-γ, or TNF-α (Giulian and Baker, 1985; Giulian et al., 1986; Sawada et al., 1989, 1990). In this study, we showed rapid morphologic change of microglia within 1.5 h in the postischemic hippocampus and suggest that these activated microglia produce TNF-α.
Astrocytes also produced TNF-α, but the production of TNF-α by astrocytes lagged 1–3 days behind production by microglia in the postischemic hippocampus. This interval seen between microglial and astrocytic production of the cytokine is in agreement with in vitro results (Sawada et al., 1989).
Function of TNF-α
The role of TNF-α in ischemic brain injury has not yet been fully elucidated. TNF-α has been shown to cause oligodendrocytic damage, suggesting an important role of TNF-α in the demyelination of the multiple sclerosis model, experimental autoimmune encephalomyelitis (Robbins et al., 1987; Selmaj and Raine, 1988; Hofman et al., 1989). It activates endothelium to bring about leukocyte adherence and procoagulant processes (Pober and Cotran, 1990; Warren, 1990; Montovani et al., 1992). Administration of TNF-α in brain ischemia has been reported to produce significant neutrophil adherence and accumulation in capillaries and small blood vessels (Liu et al., 1994). Since reducing circulating neutrophil levels or their adhesion to blood vessels can significantly reduce ischemic damage, these data suggest that TNF-α contributes to increased sensitivity and risk in ischemic brain injury. However, it also may regulate other mediators essential to cell defense (Hori et al., 1989). TNF-α stimulates the expression of nerve growth factor in a variety of cell types including fibroblasts, glioblastoma cells, and astrocytes (Hattori et al., 1993), which may contribute to neuronal survival in and around the ischemic site. TNF-α induces manganese superoxide dismutase in a variety of cells including neurons; the enzyme may protect against ischemic neuronal injury by reducing reactive oxygen intermediates (Matsuyama et al., 1994b). These findings may explain the target cell-dependent function of TNF-α in the pathophysiology of brain injury.
Both astrocytes and microglia produce TNF-α; however, this study showed that their spatial and temporal expression was different. It remains unclear whether TNF-α produced by microglia has the same functions as that produced by astrocytes in ischemic neuronal injury. TNF-α may be both a trigger and a component of the documented cytokine cascade (Abbas et al., 1991). We postulate that the following sequence of events may occur in delayed neuronal death in the hippocampus following transient global ischemia. Ischemic insult triggers microglia to secrete TNF-α, which activates astrocytes to produce other cytokines such as interleukin-6 and granulocyte-monocyte colony-stimulating factor. Subsequent production of TNF-α by activated astrocytes may facilitate neuronal injury or survival of CA1. The microglia accumulating in the injured pyramidal layer are activated and may initiate phagocytosis with TNF-α production. Recent studies have shown glial contribution to excitotoxic neuronal cell death in the hippocampus (Tsirka et al., 1995). Some proteases such as tissue plasminogen activator produced by microglia have been proposed as mediators of apoptotic cell death in the hippocampus (Gehrmann et al., 1995). TNF-α also is known to induce apoptosis through its receptor (Laster et al., 1988; Cleveland and Ihle, 1995). Thus, the later-expressed TNF-α but not the earlier-expressed one may contribute to cell death in CA1 following the transient cerebral ischemia.
In conclusion, we have demonstrated in vivo that glial cells contribute to TNF-α synthesis following cerebral ischemia. Induction of TNF-α in microglia occurs rapidly and operates at long distance, suggesting the involvement of fast and remote signaling mechanisms. In contrast to microglia, astrocytes expressed TNF-α only later when they visibly proliferated to surround the advanced ischemic lesion. It may be very difficult to elucidate whether TNF-α is beneficial or harmful in ischemic brain injury, because its action differs from cell to cell. The capacity for glial cells to react to injury by expressing the cytokine appears to be time restricted, a factor that must be considered in future neuroimmunologic experiments studying cellular mechanisms involved in ischemic brain injury.
Footnotes
Acknowledgement
The authors thank Ms. Kyoko Obata for her excellent technical assistance and for preparing the manuscript.