
Abstract
We recently showed that intraischemic moderate hypothermia (30°C) reduces ischemic damage through the Akt pathway after permanent distal middle cerebral artery occlusion in rats. The only Akt pathway component preserved by hypothermia is phosphorylated phosphatase and tensin homolog deleted on chromosome 10 (p-PTEN), which suggests that p-PTEN may have a central role in neuroprotection. Reactive oxygen species (ROS) are critically involved in mediating ischemic damage after stroke by interacting with signaling molecules, including Akt, PTEN, and δ-protein kinase C (PKC). We investigated the protective mechanisms of moderate hypothermia on these signaling proteins after transient focal ischemia in rats. Early moderate hypothermia (3h) was administered 15 mins before reperfusion, and delayed moderate hypothermia (3h) was applied 15 mins after reperfusion. Our results indicate that early hypothermia reduced infarction, whereas delayed hypothermia did not. However, both early and delayed hypothermia maintained levels of Mn-SOD (superoxide dismutase) and phosphorylated Akt and blocked δ-PKC cleavage, suggesting that these factors may not be critical to the protection of hypothermia. Nevertheless, early hypothermia preserved p-PTEN levels after reperfusion, whereas delayed hypothermia did not. Furthermore, ROS inhibition maintained levels of p-PTEN after stroke. Together, these findings suggest that phosphorylation levels of PTEN are closely associated with the protective effect of early hypothermia against stroke.
Introduction
Mild-to-moderate hypothermia is a robust neuroprotectant against cerebral ischemia (Mcilvoy, 2005; Zhao et al, 2005). Despite extensive research in the past decades, the underlying protective mechanisms of hypothermia are not fully understood. Most previous studies have concentrated on investigating the mechanisms of intraischemic hypothermia (Mcilvoy, 2005; Schwab, 2005); whereas fewer studies were conducted on postischemic hypothermia, a neuroprotectant more relevant to clinical translation.
Multiple cell signaling pathways are involved in the protective mechanism of hypothermia, including the superoxide dismutase (SOD) detoxifying system, Akt survival signaling pathway, and protein kinase C (PKC) pathways. These pathways may be regulated by reactive oxygen species (ROS) activity. For example, ROS reduce SOD activity (Chan et al, 1993; Murakami et al, 1998) and also cause proapoptotic δ-PKC, to translocate from the cytosol to the mitochondria where free radicals form, thus mediating cellular apoptosis (Churchill and Szweda, 2005; Shimohata et al, 2007). In addition, ROS regulate Akt activity (Omori et al, 2002; Lee et al, 2004) and phosphatase and tensin homolog deleted on chromosome 10 (PTEN) (Downes et al, 2004). Both Akt and PTEN are two critical molecules in the Akt pathway, in which Akt is phosphorylated, thus activated in response to growth factors. Akt blocks apoptosis by phosphorylating proapoptotic substrates, such as Bim and glycogen synthase kinase 3 beta (GSK3beta) (Zhao et al, 2005). In addition, Akt phosphorylation is negatively regulated by PTEN (Zhao et al, 2006b).
We have previously reported that 2 h of intraischemic mild hypothermia (33°C) did not offer protection in a focal ischemic model of permanent distal middle cerebral artery occlusion (MCAo) combined with 2 h of transient bilateral common carotid artery occlusion, but 4 h of mild hypothermia (2-h intraischemic plus 2-h postischemic hypothermia) reduced infarction (Zhao et al, 2007b) In addition, moderate intraischemic hypothermia (30°C) robustly reduced infarction in a permanent distal MCAo combined with 1 or 2 h of transient bilateral common carotid artery occlusion model (Zhao et al, 2005, 2007b). However, we have yet to study the protective effect of postischemic hypothermia in a transient distal MCAo model. Regarding the underlying protective mechanisms of intraischemic hypothermia, we have previously reported that hypothermia inhibits cytosolic cytochrome c release and nuclear apoptosis inducing factor translocation (Zhao et al, 2007b). In addition, we and others have shown that intraischemic hypothermia blunts the generation of free radicals, but does not alter changes in SOD protein expression after focal ischemia in rats (Maier et al, 2002). We have also found that intraischemic hypothermia reduces infarct volume by enhancing the Akt signaling pathway in a focal ischemic model (Zhao et al, 2005). Furthermore, intraischemic hypothermia blocks δ-PKC cleavage that causes apoptosis (Shimohata et al, 2007).
In this study, after defining a therapeutic time window of moderate hypothermia in a transient distal MCAo model in rats, we compared the protective effects of early with delayed hypothermia on ROS generation, protein levels of Mn-SOD, δ-PKC, p-Akt, and p-PTEN. We then investigated a potential relationship between ROS generation and decreases in p-PTEN in ischemic damage by using a ROS scavenger.
Material and methods
All animal surgery protocols were performed in accordance with the Guidelines provided by the Animal Care Committee of the Stanford University Administrative Panel.
Focal Cerebral Ischemia in Rats
Focal stroke in male Sprague—Dawley rats (280 to 330 g) was generated as previously described (Zhao et al, 2003, 2004), except that in this study, the distal MCA was occluded transiently, whereas it was occluded permanently in previous studies. The rectal body temperature was maintained by a thermostatically regulated heating pad while the rat was under anesthesia of 2% to 3% isoflurane. The distal MCA was exposed and clipped with an arterial clip (Sundt AVM Microclip No. 2, Codman, Raynham, MA, USA), and bilateral common carotid arteries were occluded using aneurysm clips. After 1 h of occlusion, the three vessels were released. Hypothermia was induced by spraying 100% alcohol onto the rat's body as previously described (Zhao et al, 2004, 2005). To study the effects of hypothermia on infarct size, rats were divided into four groups (Figure 1A), namely group 1, normothermia: the temperature was maintained at 37°C throughout the surgery; group 2, intraischemic hypothermia: the temperature was adjusted to 30°C before the onset of ischemia; group 3, early hypothermia: alcohol was sprayed onto the rat's body at 30 mins after ischemia onset, and the body temperature was adjusted to 30°C 15 mins before reperfusion; and group 4, delayed hypothermia: alcohol was sprayed onto the rat's body at the beginning of reperfusion, and the body temperature was adjusted to 30°C 15 mins after reperfusion. Body temperature of 30°C was reached 15 to 20 mins after alcohol was sprayed on the rat's body for hypothermia and maintained for 3 h. Body temperature was returned to 37°C using a light and heating pad for 30 mins under anesthesia.
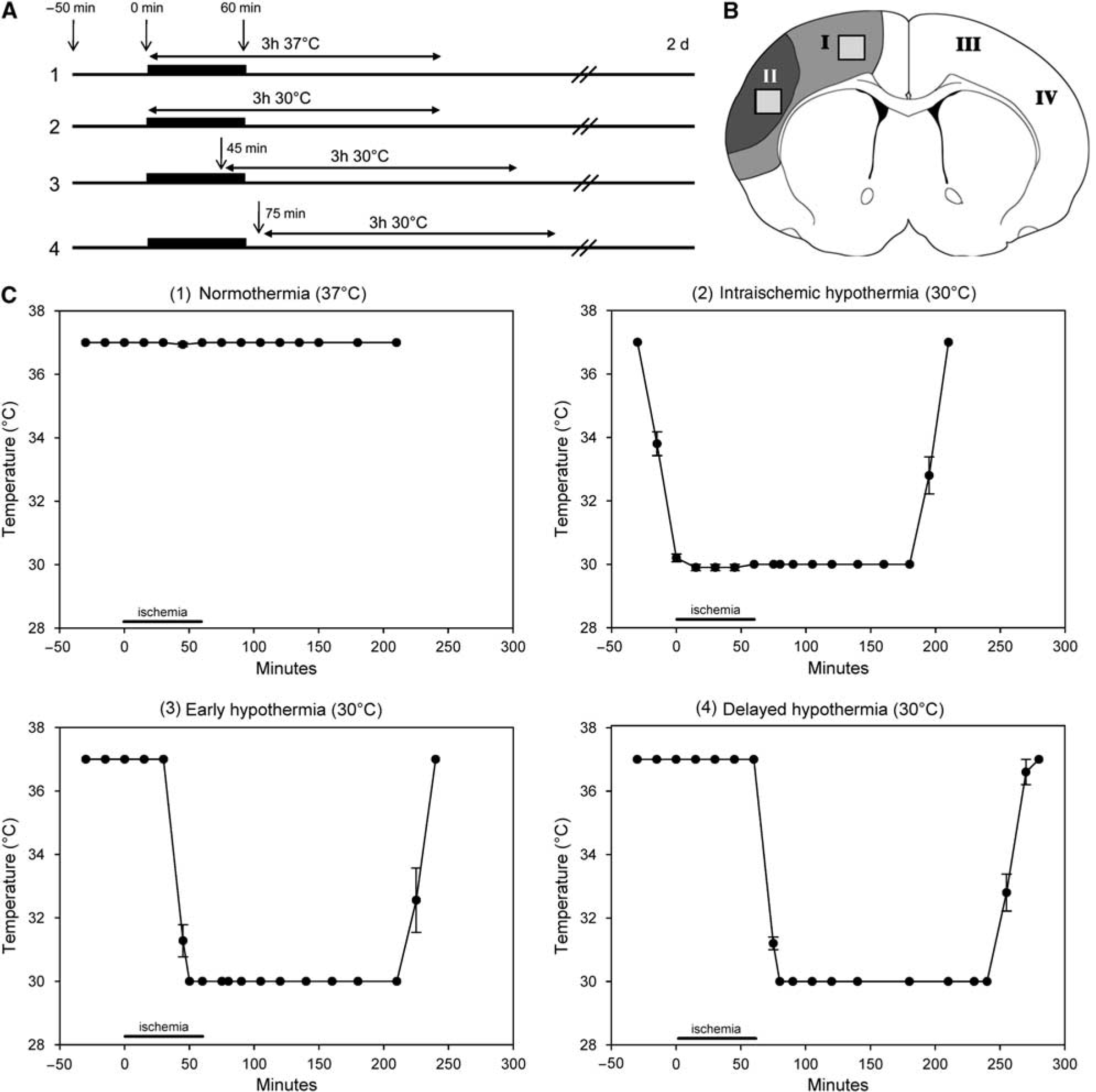
Diagrams for surgery protocols, a schematic for tissue dissection, and graphs for time courses of body temperatures. (
Analysis of Infarct Volume
For infarct volume analysis, rats were euthanized 2 days after reperfusion (
Terminal Deoxynucleotidyl Transferase-Mediated 2′-Deoxyuridine-5′Triphosphate-Biotin Nick End Labeling Staining
To investigate cell death after ischemia, rats were killed 24 h after reperfusion. Rats (
In Situ Detection of Superoxide Radicals
To detect superoxide radicals generated after ischemia, rats were injected intravenously with dihydroethidine (HEt) solution (100 μL; 1 mg/mL in PBS; Molecular Probes, Eugene, OR, USA) under anesthesia 30 mins before reperfusion as previously described (Maier et al, 2002; Zhao et al, 2006a). Rats were killed 30 mins after reperfusion followed by transcardial perfusion with heparinized PBS and 4% PFA (
Western Blot Analysis
Rats were euthanized 10, 20 mins, and 1, 3, 6, 12, and 24 h after reperfusion by an overdose of isoflurane, and transcardially perfused with cold PBS (
For total protein preparation, brain tissues were homogenized in 10 volumes of cold 1 × lysis buffer (Cell Signaling Technology, Beverly, MA, USA), including phosphatase inhibitor (Sigma), protease inhibitor (Sigma), and phenylmethylsulphonyl fluoride (1 mmol/L, Sigma) using a Dounce tissue homogenizer (Wheaton, Millville, NJ, USA). The homogenate was centrifuged at 14,000 r.p.m. for 20 mins and the supernatant was collected. Protein concentration was determined using the BCA (bicinchoninic acid) protein assay reagent (Pierce, Rockford, IL, USA). We loaded 20-μg protein onto 4% to 15% SDS-PAGE Ready Gel (Bio-Rad Laboratories, Hercules, CA, USA). After electrophoresis, the proteins were transferred onto a PVDF (polyvinylidene fluoride) membrane (Amersham Bioscience, Arlington Heights, IL, USA), and the membrane was blocked with 5% nonfat dry milk (Bio-Rad Laboratories) in PBS/0.05% Tween-20. Primary antibodies (p-PTEN, PTEN, and p-Akt, Cell Signaling Technology; δ-PKC, Santa Cruz Biotechnologies, Santa Cruz, CA, USA; 1:1,000 dilution) were incubated overnight at 4°C followed by a horseradish peroxidase (HRP)-conjugated secondary antibody (1:2,000, Cell Signaling Technology) for 1 h. Protein bands were detected using an enhanced chemiluminescence system (Amersham Bioscience) and exposed to X-ray film (Kodak, Rochester, NY, USA). The membrane was then stripped (Stripping Buffer, Amersham Bioscience) and incubated with anti-β-actin antibodies (1:5,000; Sigma) as an even-loading control of total protein.
Mitochondrial subcellular fractions were prepared using a mitochondria isolation kit (MITO-ISO1, Sigma) according to the manufacturer's instructions. Mitochondrial protein of 1 μg was subjected to 15% SDS-PAGE. After detection of Mn-SOD (1:1,000; Upstate Biotechnology, Billerica, MA, USA), the membrane was stripped to detect cytochrome oxidase subunit IV (COX IV) (Molecular Probes; 1:1,000 dilution), a marker for mitochondria. Films were scanned and the optical densities of protein bands were measured using the NIH Image J software.
Immunostaining
For immunostaining, rats were killed 24 h after reperfusion by perfusing with iced PBS followed by 4% PFA (
Free-floating immunostaining was performed as previously described (Zhao et al, 2005). For double staining of p-PTEN and Het, Het-treated brain slices were incubated at 4°C overnight with p-PTEN primary antibody (1:100 dilution, Cell Signaling Technology) diluted in a blocking solution of 5% goat serum and 0.1% Triton X-100. The tissues were then incubated with a secondary donkey antirabbit IgG (immunoglobulin G) antibody conjugated with FITC (1:200 dilution; Jackson Immuno Research Laboratories, West Grove, PA, USA). For double staining of p-PTEN and MAP-2, slices were first incubated with the primary p-PTEN antibody as described above. They were then incubated with primary microtubule-associated protein-2 (MAP-2) antibody (1:100 dilution; Sigma) overnight at 4°C, washed with PBS, and incubated for 2 h at room temperature in a solution of mixed secondary antibodies (FITC-conjugated antirabbit secondary antibody for p-PTEN, 1:200 and Cy3-conjugated antimouse secondary antibody for MAP-2, 1:200, Jackson Immuno Research Laboratories). After being washed, the slices were counterstained with DAPI (Vector Laboratories). Immunofluorescence was observed using the confocal microscopy (LSM510, Carl Zeiss).
N -Tert-Butyl-α-(2-Sulfophenyl) Nitrone Treatment
To detect whether ROS modulate phosphorylation of PTEN, we used the ROS scavenger,
Statistical Analysis
Data are presented as mean ± s.e.m. values. Statistical results from western blots were evaluated using two-way ANOVA (analysis of variance) with Tukey's
Results
The Effect of Hypothermia on Infarct Size, Apoptosis, and Free Radical Generation
The time courses of temperature changes in all groups were shown (Figure 1). Intraischemic and early hypothermia markedly reduced infarction after transient focal ischemia, whereas delayed hypothermia did not prevent ischemic damage (Figures 2A and 2B). Consistently, early hypothermia, but not delayed hypothermia, blocked TUNEL positive staining, a marker for apoptosis or cell death (Figure 2C). In addition, early hypothermia attenuated the generation of superoxide compared with normothermia (Figure 3).
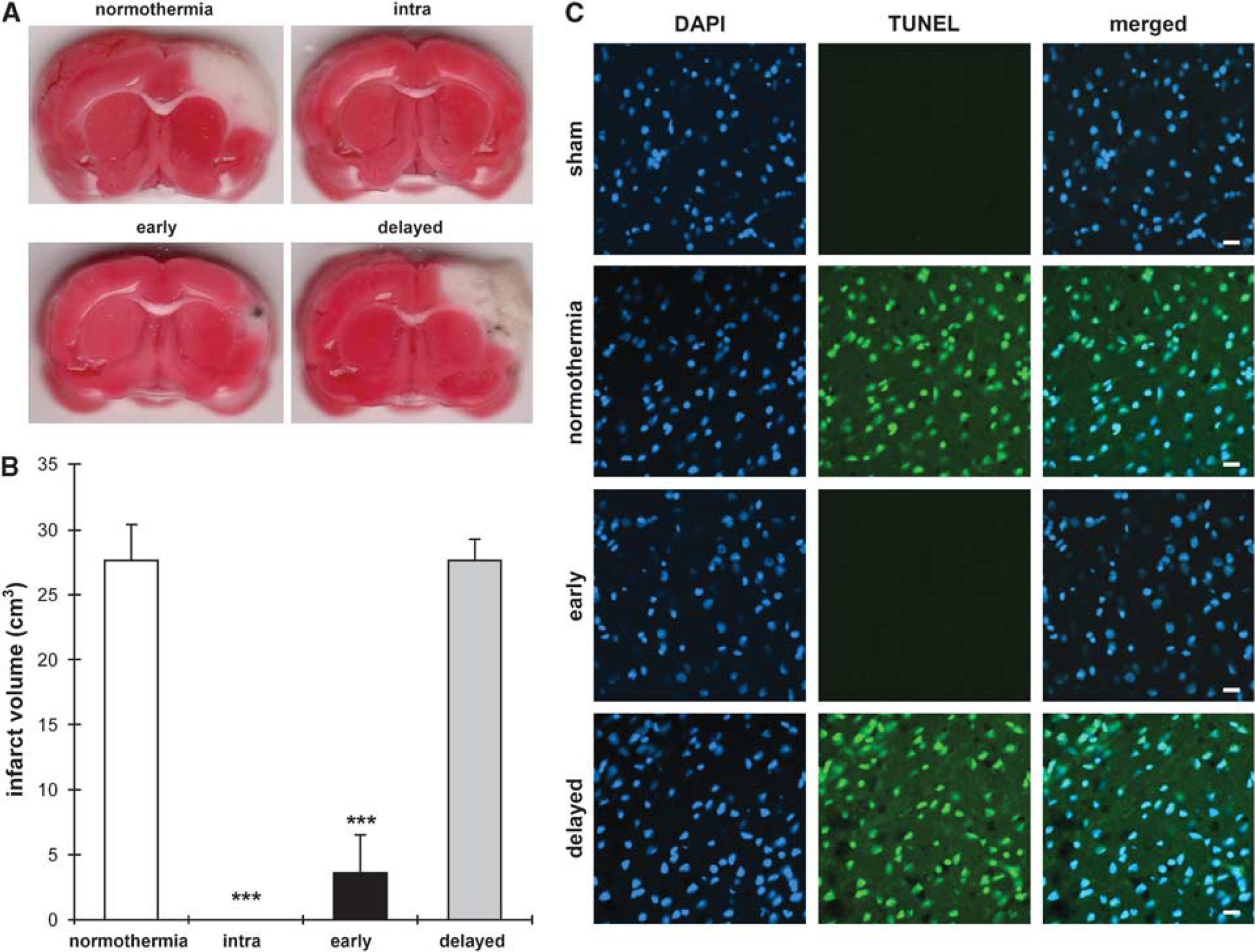
Intraischemic and early hypothermia reduce ischemic injury after transient dMCAo. (
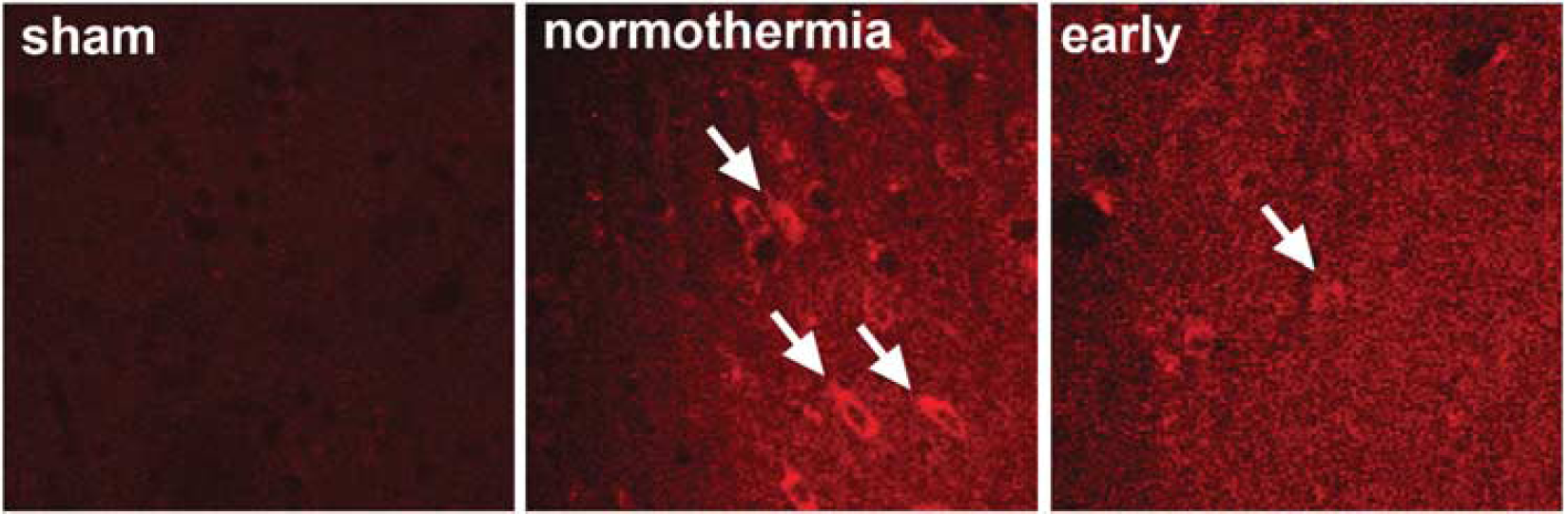
Early hypothermia blocked ROS generation in the ischemic cortex as detected by injection of dihydroethidine (HEt). Arrows indicate Et fluorescent signal derived from HEt. The study was repeated for four rats in each group. Scale bar, 20 μm.
Early and Delayed Hypothermia Showed Similar Effects on Mn-SOD Protein Levels and δ-PKC Cleavage in the Ischemic Penumbra
To investigate the underlying molecular mechanisms responsible for the protective effect of early hypothermia, we first examined the level of Mn-SOD protein at 20 mins, 1 and 3 h after reperfusion. Western blot analysis showed that normothermic stroke caused a significant reduction in Mn-SOD protein expression at 20 mins to 3 h after reperfusion in the ischemic core (Figure 4A). Early hypothermia significantly attenuated its reduction at 20 mins and 1 h in the core compared with normothermia. However, there were no differences in Mn-SOD expression levels in the penumbra between all three groups.
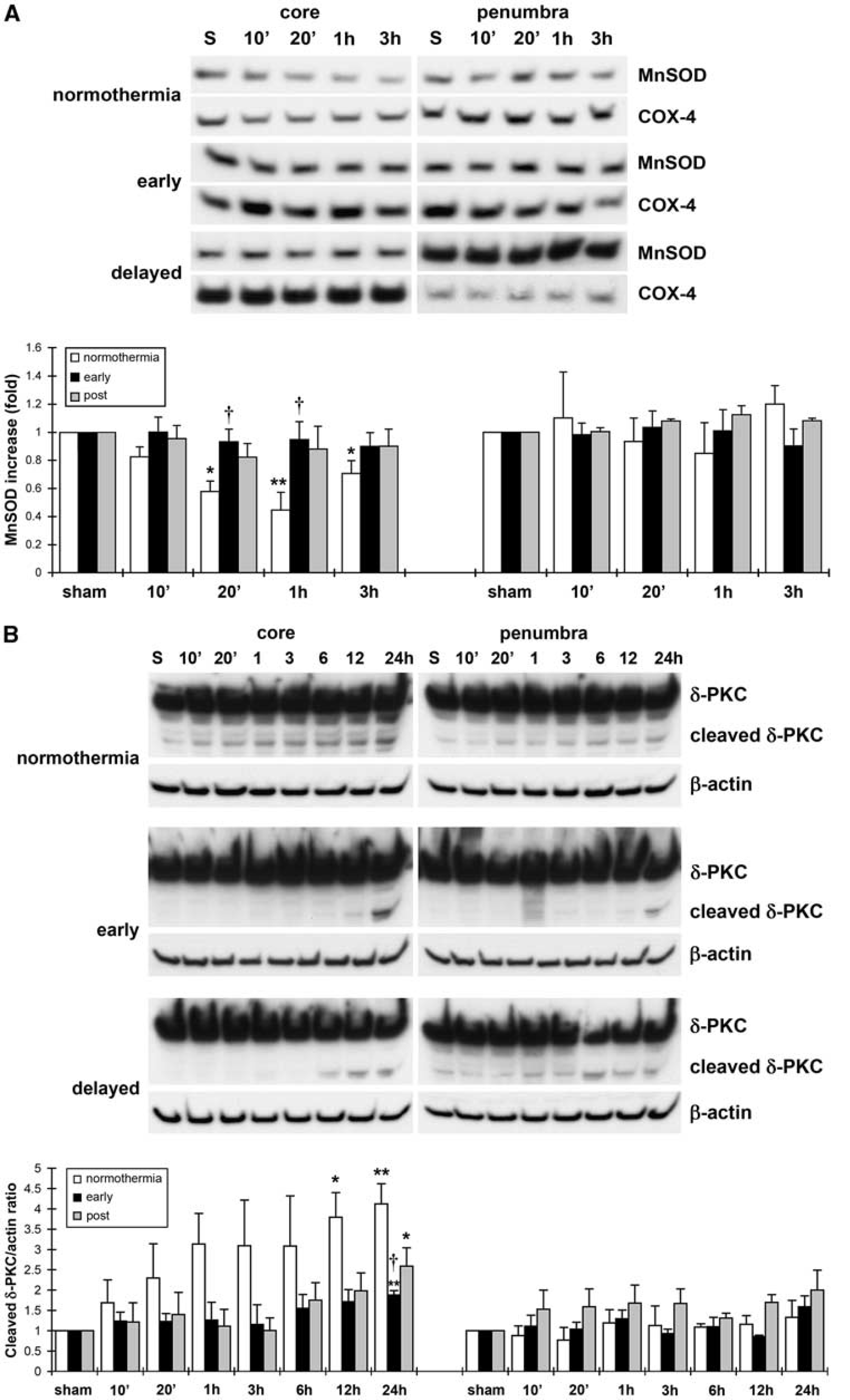
The effects of delayed hypothermia on Mn-SOD and cleaved δ-PKC. (
We also examined whether δ-PKC is involved in the protective effect of early hypothermia, as we previously reported that δ-PKC might be involved in the protective effects of intraischemic hypothermia (Shimohata et al, 2007). The cleaved form of δ-PKC increased markedly after reperfusion in the core with normothermic stroke (Figure 4B). Both early and delayed hypothermia significantly reduced δ-PKC cleavage at 24 h after reperfusion. However, there were no differences in δ-PKC cleavage in the penumbra between all three groups.
PTEN Phosphorylation was Maintained only by Early Hypothermia, Whereas Akt Phosphorylation was Preserved by Both Early and Delayed Hypothermia
We then investigated the effects of hypothermia on phosphorylation of Akt and PTEN in the Akt pathway. P-Akt levels decreased at 12 and 24 h in the core, which was blocked by both early and delayed hypothermia (Figure 5). In the penumbra, p-Akt significantly increased at 1 h and decreased at 24 h in normothermic brains; both early and delayed hypothermia appear to block these changes. However, there was no statistically significant difference between groups with early and delayed hypothermia.
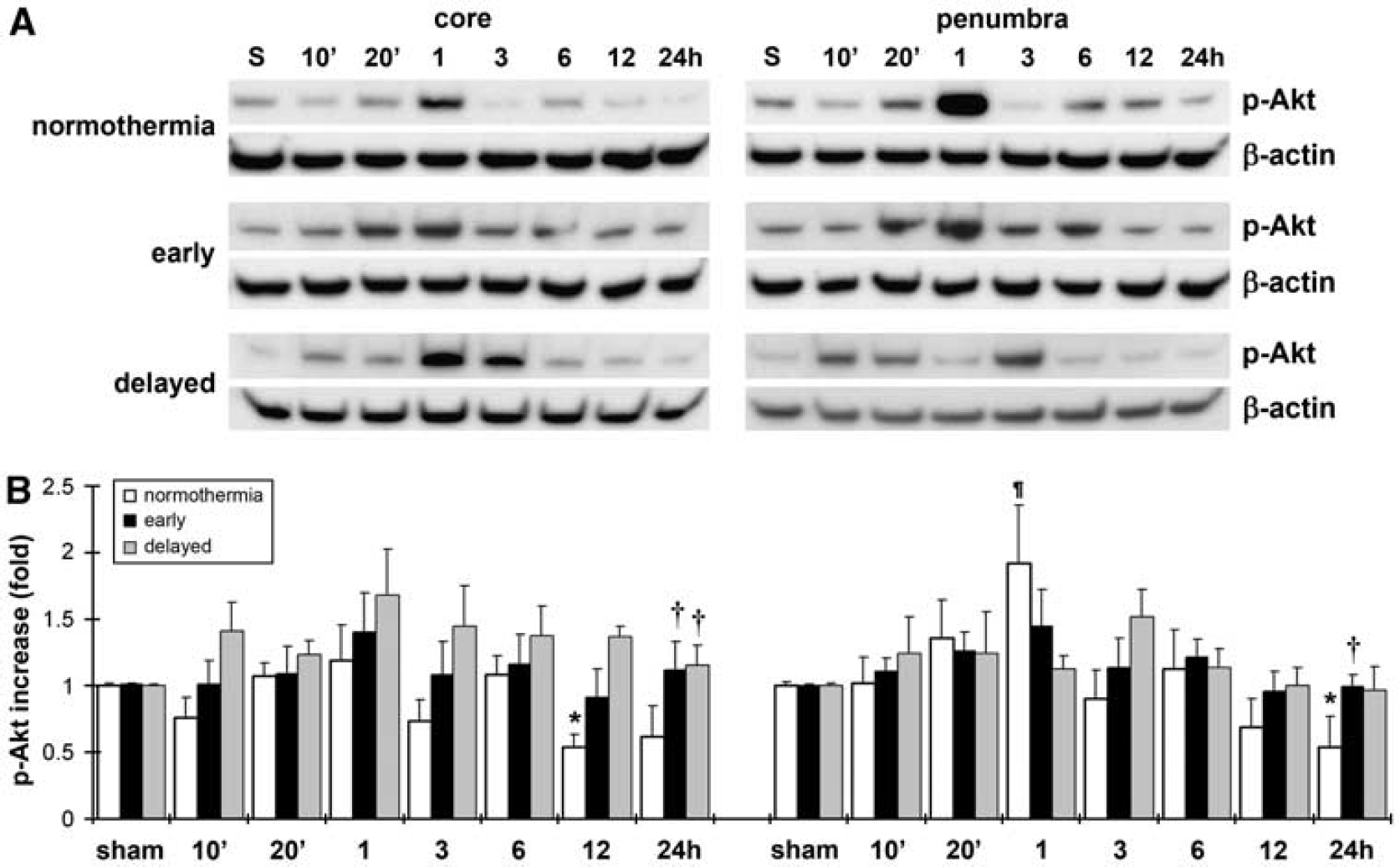
Effects of hypothermia on p-Akt. (
Phosphorylated phosphatase and tensin homolog deleted on chromosome 10 levels rapidly decreased in the core and penumbra at all time intervals after reperfusion (Figures 6A and 6B). Early, but not delayed hypothermia, preserved p-PTEN expression at 6 and 24 h after reperfusion in the core and at 1, 3, 6, 12, and 24 h in the penumbra.
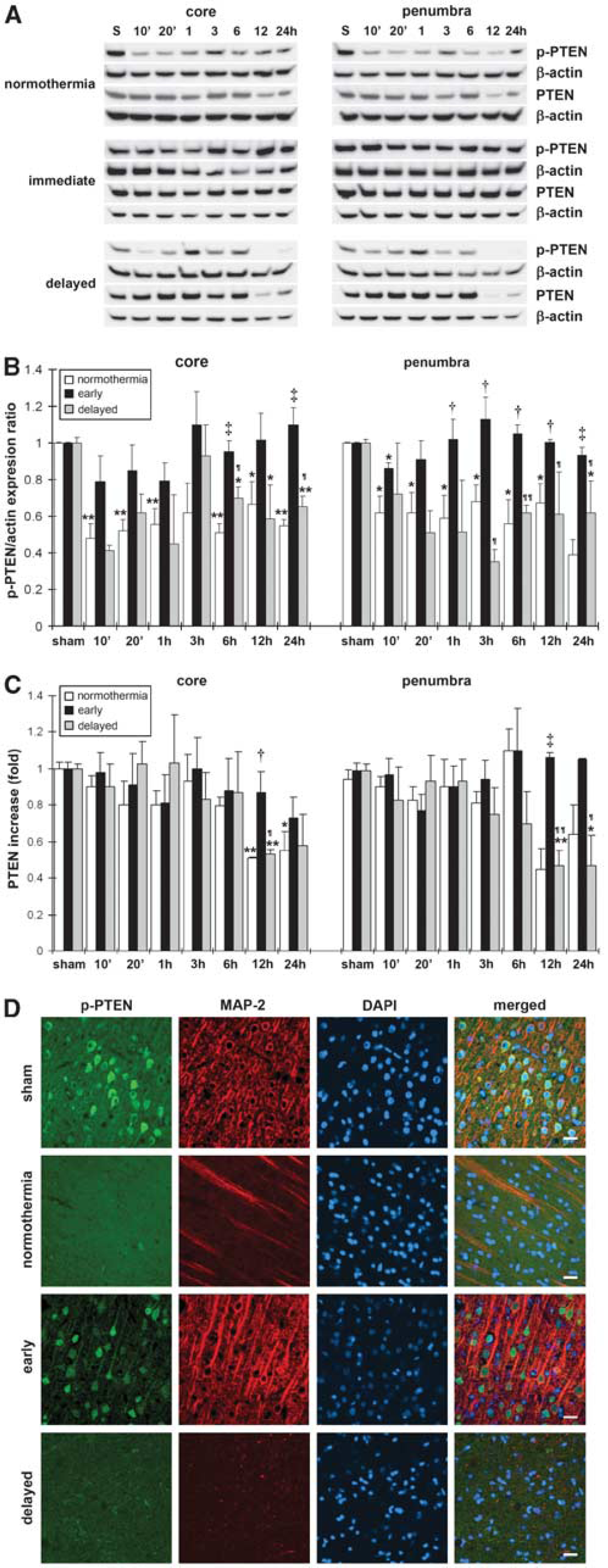
Early, but not delayed hypothermia preserve protein levels of p-PTEN and PTEN after reperfusion. (
To investigate whether the p-PTEN expression correlated with neuroprotection in early hypothermia, double immunostaining of p-PTEN and MAP-2, a marker for neuronal survival, was performed. The results showed that p-PTEN colocalized with MAP-2 in the brain, and that the p-PTEN expression was decreased in normothermic and delayed hypothermic brains, but was preserved in early hypothermic brains (Figure 6D).
Normothermic stroke also caused decreases in PTEN protein levels at 12 and 24 h after reperfusion in the core and penumbra. Early, but not delayed hypothermia maintained PTEN protein levels at 12 h in the core and penumbra compared with normothermia (Figure 6C).
Reactive Oxygen Species Might Reduce p-PTEN Expression After Stroke
We next investigated whether ROS were involved in the reduction of p-PTEN after reperfusion. Immunostaining showed that ischemic neurons with intense Et staining in the ischemic brain treated with normothermia correspond to reduced p-PTEN expression (Figure 7A), whereas Et staining was not detected in ischemic brains with early hypothermia. We then investigated whether S-PBN, a ROS scavenger, blocked the reduction in p-PTEN levels. We found that S-PBN injected at ischemic onset reduced infarct volume by 57.2% (Figure 7B). Western blots indicated that S-PBN significantly inhibited decreases in p-PTEN protein level in the penumbra 30mins after reperfusion (Figure 7C).
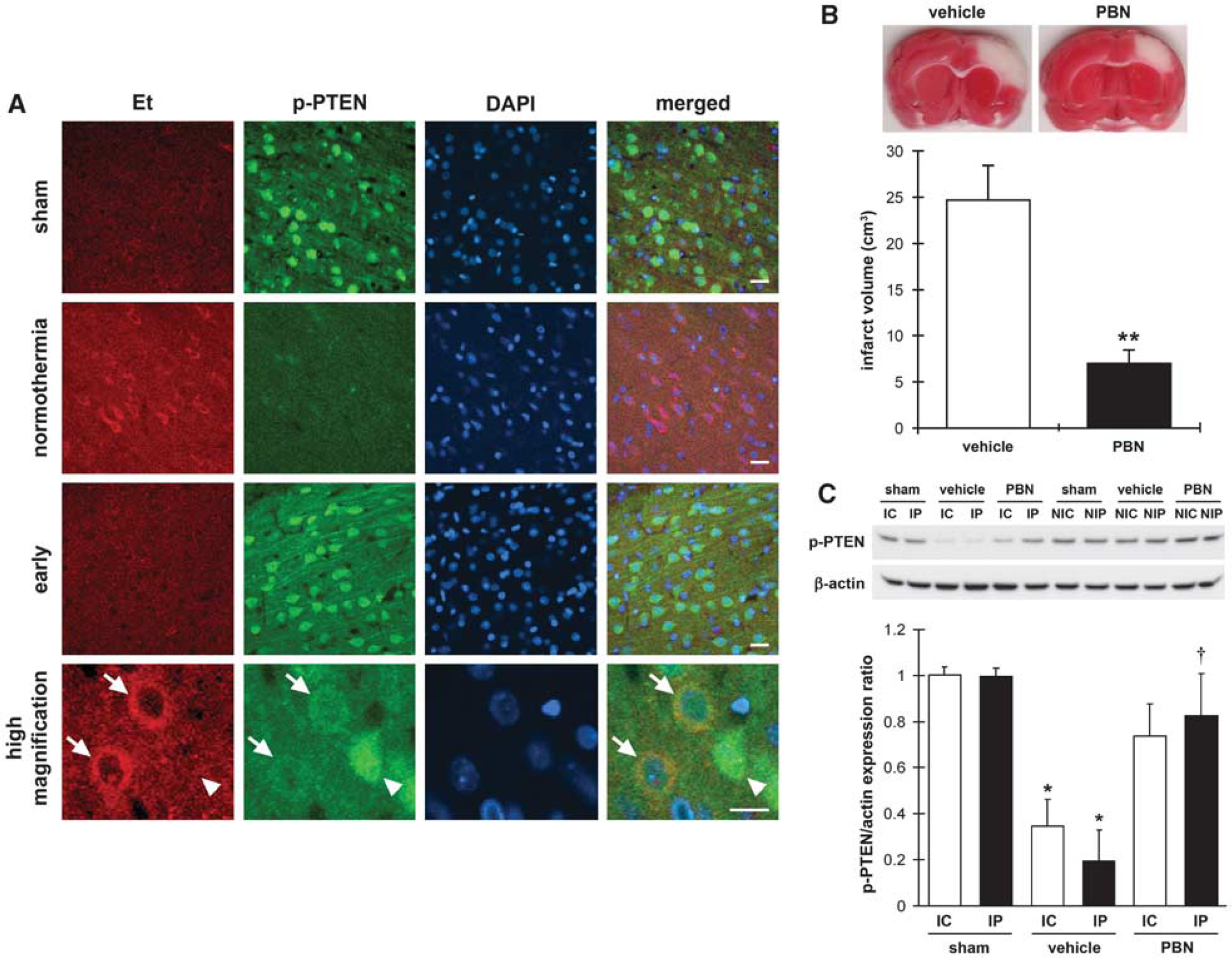
(
Discussion
In this study, we found that a 15- to 20-min delay in hypothermia application (delayed hypothermia) failed to reduce infarct volume compared with hypothermia initiated 15 mins before reperfusion (early hypothermia). It appears that the protective effect of early hypothermia was achieved in part by its ability to reduce free radical generation. However, this effect is not associated with SOD2 protein expression or δ-PKC cleavage, because no significant difference in their expression was found between ischemic brains treated with early or delayed hypothermia. Instead, the protective effect of early hypothermia correlates with its effect on p-PTEN, as both early hypothermia and a free radical scavenger reduced infarct size and preserved p-PTEN levels after stroke.
As we recently reviewed (Zhao et al, 2007a), the therapeutic efficacy of postischemic hypothermia depends on the time of onset, duration, and depth of hypothermia. Previous studies have shown the protective effect of postischemic hypothermia when it was maintained for 1 to 3 h after MCAo in adult rats (Chen et al, 1992; Xue et al, 1992, Zhang et al, 1993a, b ; Markarian et al, 1996). In these studies, hypothermia was initiated at various time points after ischemic onset, from as early as 15 mins after ischemic onset (Xue et al, 1992) to 1 h after reperfusion (Zhang et al, 1993b). However, the therapeutic time window must be coordinated with a hypothermic duration to protect against stroke. For example, Ohta et al (2007) suggested that the therapeutic time window of mild hypothermia was 4 h after reperfusion when it was maintained for 2 days in rats subjected to 2 h of MCAo. However, when the duration of hypothermia was shortened to 24 h, even when it was applied immediately after stroke, hypothermia offered no protection against stroke. In our study, we have defined a narrow therapeutic time window for moderate hypothermia (3 h) in a transient distal MCAo model in rats; hypothermia must be instituted before the onset of reperfusion, whereas hypothermia initiated immediately after reperfusion eliminated the protective effects of hypothermia. Our observation of a short therapeutic time window is not a novel idea. Yanamoto et al (1996) had shown that immediate hypothermia maintained for 21 h significantly reduced infarction; however, it provided no protection when hypothermia was delayed for 30 mins. More recently, Nowak's group have shown that hypothermia (32°C, 2 h) initiated 5 mins before reperfusion offered no protection in a 90-min transient focal ischemic model in spontaneously hypertensive rats (Kurasako et al, 2007). However, hypothermia (32°C) initiated at 30 mins of occlusion (60 mins before reperfusion) provided nearly complete protection (Ren et al, 2004) Thus, our study concurs with these previous studies, suggesting that hypothermia must be initiated before reperfusion onset to achieve neuroprotection, and a 20- to 30-min delay of hypothermic application failed to provide hypothermic protection. Thus, the onset time and duration of hypothermia must be carefully determined before clinical translation. Nevertheless, it is inappropriate to directly extrapolate these experimental findings to clinical translation, for the protective effect of hypothermia is species or strain dependent (Kurasako et al, 2007); thus, whether similar therapeutic time windows exist for human stroke patients is not known. Despite this, our study, together with other previous reports, serves as a reminder that hypothermia must be instituted as early as possible for stroke treatment.
We then explored the underlying protective mechanisms of early hypothermia. As ROS are believed to be an initiator and a mediator of cell death pathways in reperfusion injury (Sugawara and Chan, 2003; Fujimura et al, 2005), we first examined free radical products 30 mins after reperfusion, and found that early hypothermia reduced ROS generation. Previous reports, including ours, have suggested that both intraischemic hypothermia (Globus et al, 1995; Kil et al, 1996; Maier et al, 2002) and postischemic hypothermia (Ishikawa et al, 1999; Kawai et al, 2000) reduce ROS generation after reperfusion. Thus, it is plausible that inhibiting ROS activity contributes to the protective effect of early hypothermia.
The effect of early hypothermia on ROS does not correlate with its effect on SODs and δ-PKC, as no significant differences in their protein levels were detected in between early and delayed hypothermic brains. It is well known that SOD1 (Cu/Zn-SOD) is constitutively expressed in the cytosol (Chan et al, 1993) and SOD2 (Mn-SOD) is primarily localized to the mitochondria (Murakami et al, 1998). Our preliminary data suggest that SOD1 did not change after stroke in this model (data not shown). However, SOD2 significantly decreased in the mitochondria after reperfusion in the core. Early hypothermia preserved SOD2 protein levels in the core, but there were no changes of SOD2 in the penumbra in both early and delayed hypothermia. In addition, δ-PKC facilitates apoptosis in various types of cells, including neurons (Anantharam et al, 2002; Brodie and Blumberg, 2003), and it is well known to mediate ROS effects after ischemia (Bright et al, 2004; Bright and Mochly-Rosen, 2005; Otani, 2004). Recently, we found that δ-PKC cleavage occurs after stroke and that δ-PKC inhibition contributes to the protective effect of intraischemic hypothermia (Shimohata et al, 2007). Consistent with our previous study, the cleaved form of δ-PKC after reperfusion was increased after normothermic stroke. As both early and delayed hypothermia inhibited δ-PKC cleavage in the ischemic core, where infarction is irreversible, inhibition of δ-PKC cleavage does not correlate with infarct reduction. In addition, significant changes in Mn-SOD and δ-PKC were not detected in the ischemic penumbra, where early hypothermia was protective without affecting their protein levels. Therefore, we conclude that the preservation of SOD2 protein and inhibition of δ-PKC cleavage do not have major roles in the protective effect of early hypothermia.
Nevertheless, the protective effect of early hypothermia is associated with its ability in maintaining p-PTEN levels, which also seems to correlate with ROS inhibition on the basis of our observations that early hypothermia blocked ROS generation, both early hypothermia and the ROS scavenger reduced infarct size and preserved p-PTEN, and that the p-PTEN expression did not colocalize with superoxide products. This protective effect is consistent with our previous study showing that intraischemic hypothermia protects the ischemic brain from damage by preserving p-PTEN (Zhao et al, 2005). In general, PTEN activity is regulated by the balance of its phosphorylation and dephosphorylation (Torres and Pulio, 2001; Vazquez et al, 2001). Dephosphorylated PTEN is active, but degraded rapidly under normal conditions (Georgescu et al, 2000; Das et al, 2003). In our study, PTEN is dephosphorylated early in reperfusion; early hypothermia, but not delayed hypothermia, maintains PTEN stability by preserving its phosphorylation. Although several papers have suggested that PTEN is involved in the pathologic process in cerebral ischemia (Majumder et al, 2001; Omori et al, 2002; Lee et al, 2004; Ning et al, 2004), little is known about the pathologic roles of PTEN and the underlying ROS-dependent signaling processes. For the first time, we found that S-PBN, a free radical scavenger, reduces infarct size and preserves p-PTEN protein level, providing evidence that ROS activity might be partially responsible for decreases in p-PTEN after stroke.
Our study has some limitations. First, the Het signals representing ROS activity were evaluated at only one time point, i.e., 30 mins after recirculation. It would be inappropriate to study the effect of delayed hypothermia at this time point because that delayed hypothermia was only adjusted to the targeted temperature 15 mins before this time point, which might provide insufficient time for it to inhibit ROS activity. Nevertheless, we have no evidence whether delayed hypothermia inhibited ROS activity at later time points. A similar limitation exists in the study of double staining of Het and p-PTEN (Figure 7), which was also carried out on brain tissues harvested at 30 mins after reperfusion. Second, ROS and p-PTEN expression were not analyzed at multiple sites on the ischemic cortex, and ROS activity was not measured quantitatively. Third, although we are tempted to use the effect of S-PBN on p-PTEN as evidence supporting that the effect of hypothermia on p-PTEN preservation was due to its ability to block ROS activity, more work is needed to address whether the protective mechanisms of hypothermia is comparable or similar with those of S-PBN. All of these limitations may weaken our initial experimental purposes regarding the protective effects of hypothermia and S-PBN on ROS activity, as well as the associated p-PTEN and other protein expressions.
In conclusion, early hypothermia appears to reduce infarct size by blunting free radical generation after reperfusion. PTEN, but not δ-PKC or SOD2, may have a critical role in mediating the protective effect of early hypothermia. This study may lead to a deeper understanding of the precise role of ROS in causing ischemic injury, and help identify novel therapeutic targets for treating stroke.
Footnotes
Acknowledgements
The authors deeply appreciate David Kunis for technical assistance, Beth Hoyte for preparation of the figures, and Felicia Beppu for manuscript preparation.
The authors declare no conflict of interest.