
Abstract
The blood—retinal barrier (BRB) is essential for the normal structural and functional integrity of the retina, whose breakdown could cause the serious vision loss. Vascular endothelial growth factor (VEGF), as a permeable factor, induces alteration of tight junction proteins to result in BRB breakdown. Herein, we demonstrated that decursin inhibits VEGF-mediated inner BRB breakdown through suppression of VEGFR-2 signaling pathway. In retinal endothelial cells, decursin inhibited VEGF-mediated hyperpermeability. Decursin prevented VEGF-mediated loss of tight junction proteins including zonula occludens-1 (ZO-1), ZO-2, and occludin in retinal endothelial cells, which was also supported by restoration of tight junction proteins in intercellular junction. In addition, decursin significantly inhibited VEGF-mediated vascular leakage from retinal vessels, which was accompanied by prevention of loss of tight junction proteins in retinal vessels. Decursin significantly suppressed VEGF-induced VEGFR-2 phosphrylation that consequently led to inhibition of extracellular signal-regulated kinase (ERK) 1/2 activation. Moreover, decursin induced no cytotoxicity to retinal endothelial cells and no retinal toxicity under therapeutic concentrations. Therefore, our results suggest that decursin prevents VEGF-mediated BRB breakdown through blocking of loss of tight junction proteins, which might be regulated by suppression of VEGFR-2 activation. As a novel inhibitor to BRB breakdown, decursin could be applied to variable retinopathies with BRB breakdown.
Keywords
Introduction
The blood—retinal barrier (BRB) is essential for the normal structural and functional integrity of the retina, which is formed by the endothelial cells of the retinal blood vessels (inner BRB) and the retinal pigment epithelial cells (outer BRB) (Cunha-Vaz, 1976). As the BRB serves critical functions in the eye, the BRB breakdown could therefore result in the serious vision loss (Kim et al, 2006). For example, diabetic retinopathy is a common cause of inner BRB breakdown whereas age-related macular degeneration is a common cause of outer BRB breakdown. In the retina, hypoxic—ischemic stress seems to be an important cause of BRB breakdown (Kaur et al, 2008). However, inner and outer BRB demonstrate differential sensitivities and different clinical features to hypoxic—ischemic insult. As outer BRB breakdown could result from hypoxic—ischemic insult only when choroidal circulation is generally reduced, outer BRB seems to be resistant to the hypoxicischemia (Johnson and Foulds, 1978). Therefore, ischemia-induced disruption of BRB is largely restricted to inner BRB (Koto et al, 2007). Retinal ischemia leads to inner BRB breakdown, which is characterized by vascular leakage because of increased vascular permeability (Wilson et al, 1995). Vascular endothelial growth factor (VEGF), the best known pro-angiogenic factor, is originally isolated as a vascular permeability factor to increase the vascular permeability of microvessels through uncoupling of junctional molecules in endothelial cells (Weis and Cheresh, 2005). It has been known that VEGF, sufficient to induce vascular abnormalities including vascular leakage, microangiopathy, and neovascularization (Tolentino et al, 1996), plays a major role in the initiation and development of variable retinopathies (Aiello et al, 1994). Therefore, VEGF-mediated alteration of tight junction proteins leads to BRB breakdown (Choi et al, 2007; Kim et al, 2009a).
Angelica gigas Nakai has been known as a traditional, medicinal plant in East Asia. Especially, decursin isolated from the root of A. gigas Nakai (Konoshima et al, 1968) has been reported to have variable pharmacological properties including neuroprotection (Kang and Kim, 2007), antibacterial (Lee et al, 2003a), and antitumor activities (Itokawa et al, 1994). Recently, we found out that decursin, isolated from the root of A. gigas Nakai (Konoshima et al, 1968), is a potent angiogenesis inhibitor in tumor, which effectively inhibited VEGF-induced angiogenic processes in vitro and in vivo through inactivation of VEGFR-2 signaling pathway (Jung et al, 2009).
In this study, we demonstrated that decursin inhibits VEGF-mediated inner BRB breakdown through suppression of VEGFR-2 signaling pathway. Decursin effectively suppressed VEGF-mediated hyperpermeability in retinal vascular endothelial cells, which was accompanied by blocking of VEGF-induced loss of tight junction proteins. In addition, decursin significantly inhibited VEGF-mediated vascular leakage from retinal vessels, which was closely associated with restoration of tight junction proteins in retinal vessels. The antipermeable activity of decursin was mediated by suppression of VEGF-induced VEGFR-2 activation, which was followed by the consequent inhibition of extracellular signal-regulated kinase (ERK) 1/2 activation, but not of Akt activation. Interestingly, decursin never affected on the cellular viability of retinal vascular endothelial cells and induced no retinal toxicity up to five times of its effective therapeutic concentration.
Materials and methods
Mouse
C57BL/6 mice were purchased from Samtako (Seoul, Korea). Care, use, and treatment of all animals in this study were in strict agreement with the Association for Research in Vision and Ophthalmology (ARVO) statement for the use of animals in ophthalmic and vision research. C57BL/6 mice were kept in standard 12-h dark-light cycles and approximately 23°C room temperature.
Cell culture
Human retina microvascular endothelial cells (HRMECs) were purchased from the Applied Cell Biology Research Institute (Kirkland, WA, USA) and grown on attachment factor-coated plates in complete medium (Cell Systems, Kirkland, WA, USA) or in M199 medium supplemented with 20% fetal bovine serum (FBS), 3 ng/mL basic fibroblast growth factor (Sigma, St Louis, MO, USA), and 10 U/mL heparin (Sigma). HRMECs used in this study were taken from passages 4 to 6. Decursin was extracted from the roots of A. gigas Nakai and isolated using silica gel column chromatography as our earlier report (Jung et al, 2009). Decursin or VEGF (20 ng/mL, Sigma) treatment was performed in cells cultured in serum-free M199 supplemented with 1% (vol/vol) penicillin-streptomycin.
[3H] Sucrose Permeability Assay
As our earlier description (Min et al, 2007), HRMECs (1 times 105 cells) were plated onto a Transwell filter (Corning Costar, Cambridge, MA, USA). After reaching confluence, 20 ng/mL VEGF or 10 μmol/L decursin was treated for 6 h in HRMECs; 50 μL (0.8 μCi/mL) [3H] sucrose (1 μCi/μL; Amersham Pharmacia, Bucks, UK) was added to the upper compartment. The amount of radioactivity that diffused into the lower compartment was determined after 30 mins by liquid scintillation counter (Perkin Elmer/Wallac, Gaithersburg, MD, USA).
Leakage Assessment by Perfusion of Retinal Vessels with FITC-Bovine Serum Albumin
At 24 h after the intravitreal injection of VEGF with or without 10 μmol/L decursin, deeply anesthetized mice were perfused through the tail vein with fluorescein isothiocyanate (FITC)-bovine serum albumin (FITC-BSA, Sigma). The number of mice used in each experimental group was six. For retinal flat-mounting, after 1 h perfusion, the eyes were enucleated and fixed in 4% paraformaldehyde for 2 h. The retinas were dissected, flat-mounted in Dako mounting medium (DakoCytomation, Glostrup, Denmark), and viewed by fluorescence microscopy (BX50, OLYMPUS, Tokyo, Japan). For quantification of retinal vascular leakage, after 1 h perfusion, the eyes were enucleated, embedded in optimal cutting temperature compound (OCT) medium (Sakura Finetek Europe, Zoeterwoude, the Netherlands), and immediately frozen in liquid nitrogen. The plasma was collected and assayed for fluorescence with an SPEX fluorescence spectrophotometer (Molecular Devices, Sunnyvale, CA, USA) based on standard curves of FITC-BSA in normal mouse plasma. Frozen retinal sections (4 to 8 μm thick) collected every 30 μm were viewed with a fluorescence microscope (BX50, OLYMPUS), and six images from nonvascular retina (200 μm2) in each section were collected. Quantification of FITC-BSA fluorescence intensity was calculated by computer software Q-win (Leica, Wetzlar, Germany) and normalized to plasma fluorescence intensity for each animal.
Western Blotting
Western blotting was performed using standard Western blotting methods. The protein concentration was measured using a bicinchoninic acid (BCA) protein assay kit (Pierce, Rockford, IL, USA). Equal amounts of protein were separated by electrophoresis on 5% to 10% SDS-PAGE and transferred electrophoretically onto nitrocellulose membrane (Amersham, Little Chalfont, UK). The membranes after blocking were incubated overnight with antizonula occludens-1 (ZO-1, Zymed, San Francisco, CA, USA), anti-ZO-2 (Zymed), antioccludin (Zymed), anti-VEGFR-2 (Cell Signaling Technology, Beverly, MA, USA), antiphospho-VEGFR-2 (pVEGFR-2, Cell Signaling Technology), anti-Akt (Cell Signaling Technology), antiphospho-Akt (pAkt, Cell Signaling Technology), anti-ERK 1/2 (Cell Signaling Technology), and antiphospho-ERK 1/2 (pERK 1/2, Cell Signaling Technology) at 4°C. To ensure the equal loading of protein in each lane, the blots were stripped and reprobed with an antibody against β-actin. Intensity values were normalized relative to control values. The blots were scanned using a flatbed scanner and the band intensity analyzed using the TINA software program (Raytest, Staubenhardt, Germany).
Immunocytochemistry
ZO-1 and occludin expression in intercellular junction were examined by an immunocytochemical method as our earlier description (Min et al, 2005). Briefly, treated cells were fixed with 2% paraformaldehyde and permeabilized with 0.2% Triton X-100. After being washed in phosphate buffered saline (PBS), the slides were blocked with 3% BSA for 1 h and the cells incubated with anti-ZO-1 (1:100, Zymed) and antioccludin (Zymed) at 4°C. The slides were viewed by fluorescence microscopy (BX50, OLYMPUS).
Immunohistochemistry
The enucleated mice eyes used for immunohistochemistry were immersion fixed in 4% paraformaldehyde and subsequently embedded in paraffin; 4μm-thick serial sections were prepared from paraffin blocks. Sections were deparaffinized and hydrated by sequential immersion in xylene and graded alcohol solutions, treated with proteinase K at 37°C, and then treated with normal serum obtained from the same species in which the secondary antibody was developed to block nonspecific staining. Slides were incubated overnight at 4°C with anti-ZO-1 (1:100, Zymed) and antioccludin (1:100, Zymed) with antiplatelet/endothelial cell adhesion molecule (PECAM) (Santa Cruz Biotechnology, Santa Cruz, CA, USA). The slides were viewed by fluorescence microscopy (BX50, OLYMPUS).
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium Bromide (MTT) Assay
Cellular viability was evaluated with MTT assay. HRMECs (1 times 105 cells) were plated in 96-well plates and cultured overnight, which were treated with 0.1 to 100 μmol/L decursin for 48 h. The medium was then replaced with fresh medium containing 0.5 mg/mL MTT for 4 h. After incubation, the medium was carefully removed from the plate and dimethyl sulfoxide (DMSO) was added to solubilize formazan produced from MTT by the viable cells. Absorbance was measured at 540 nm using a microplate reader (Molecular Devices).
Terminal Deoxynucleotidyl Transferase Biotin-dUTP Nick End Labeling (TUNEL) Assay
50 μmol/L decursin, five times over the effective therapeutic concentration, was intravitreally injected to 8-week-old female C57BL/6J mice. The mice were KILLED at 3 days after the injection and enucleated. Enucleated globes were fixed in 4% paraformaldehyde for 24 h, and embedded in paraffin. TUNEL staining was performed with a kit (ApopTag Fluorescein Green; Intergen, Purchase, NY, USA), according to the manufacturer's instructions. TUNEL-positive cells were evaluated in randomly selected fields at a × 400 magnification viewed under fluorescein microscopy (BX50, OLYMPUS).
Statistical Analysis
Statistical differences between groups were evaluated using either a Mann-Whitney test or a one-way analysis of variance (ANOVA) with Tukey's post hoc test using SPSS for Windows, version 12.0 (SPSS, Chicago, IL, USA). Mean ± s.d. is shown. P≤0.05 was considered significant.
Results
Decursin Inhibits VEGF-Mediated Hyperpermeability in Retinal Endothelial Cells
To evaluate antipermeable effect of decursin on retinal endothelial cells, [3H] sucrose permeability assay was conducted in HRMECs. According to our earlier report (Jung et al, 2009), 10 μmol/L decursin, the therapeutically effective concentration in in vitro and in vivo, was treated. As shown in Figure 1, [3H] sucrose permeability in HRMECs was significantly increased 2.6-fold by 20 ng/mL VEGF treatment, which was however completely inhibited by cotreatment of 10 μmol/L decursin with 20 ng/mL VEGF (P < 0.05).
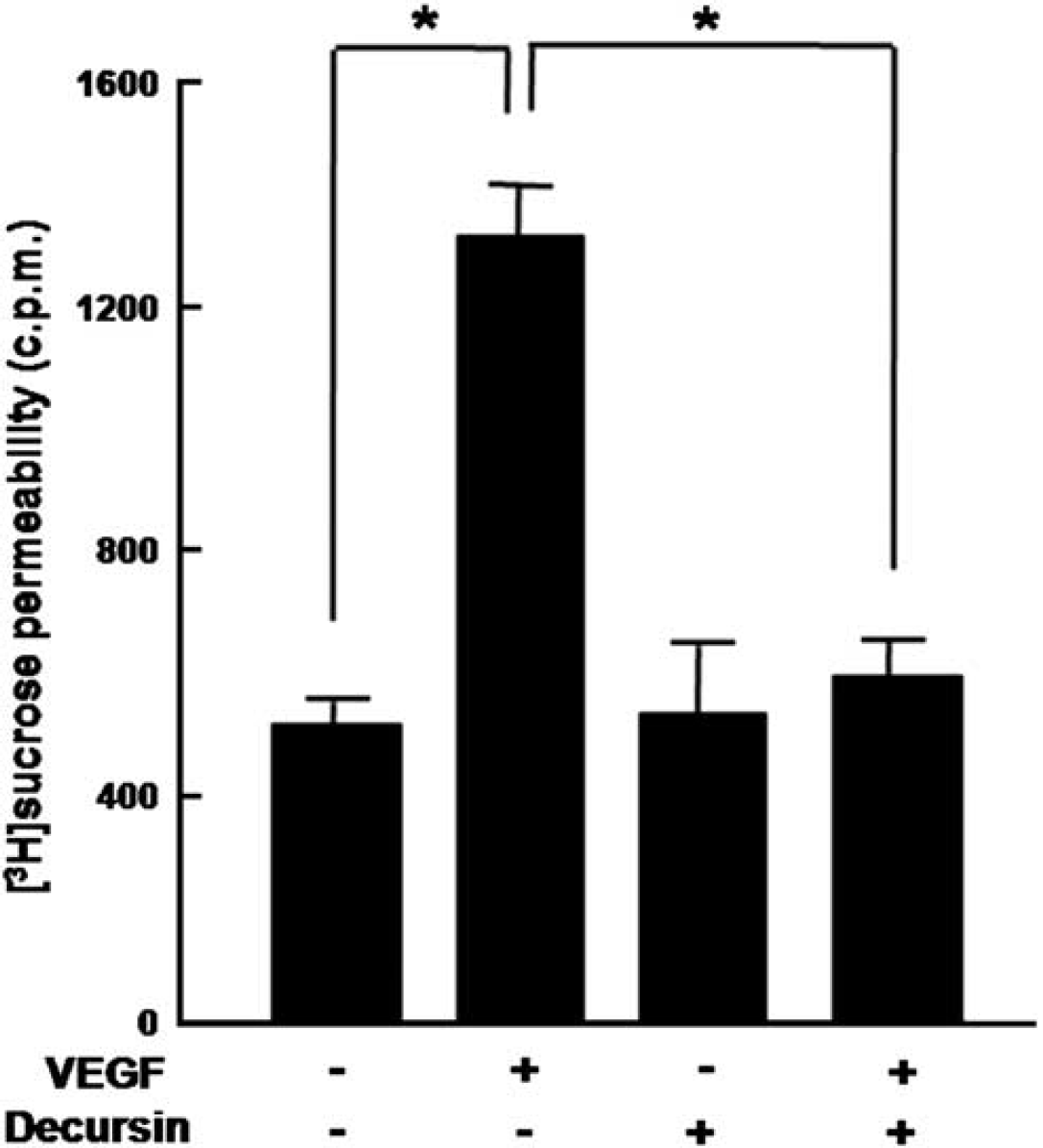
Decursin inhibits VEGF-mediated hyperpermeability in retinal endothelial cells. An amount of 20 ng/mL VEGF or 10 μmol/L decursin was treated for 6 h in HRMECs. [3H] sucrose permeability assay in HRMECs treated with VEGF or decursin was measured as counts per minute (c.p.m.). Each point represents the mean (± s.d.) of three independent experiments, each performed in triplicate. *P < 0.05.
Decursin Prevents VEGF-Mediated Loss of Tight Junction Proteins in Retinal Endothelial Cells
To investigate the preventive effect of decursin on VEGF-mediated loss of tight junction proteins in retinal endothelial cells, the expression of tight junction proteins including ZO-1, ZO-2, and occludin was assessed with or without decurin in VEGF-treated HRMECs; 20 ng/mL VEGF significantly reduced the expression of tight junction proteins such as ZO-1, ZO-2, and occludin in HRMECs, whereas 10 μmol/L decursin nearly prevented VEGF-mediated loss of tight junction proteins (Figure 2A) (P < 0.05).
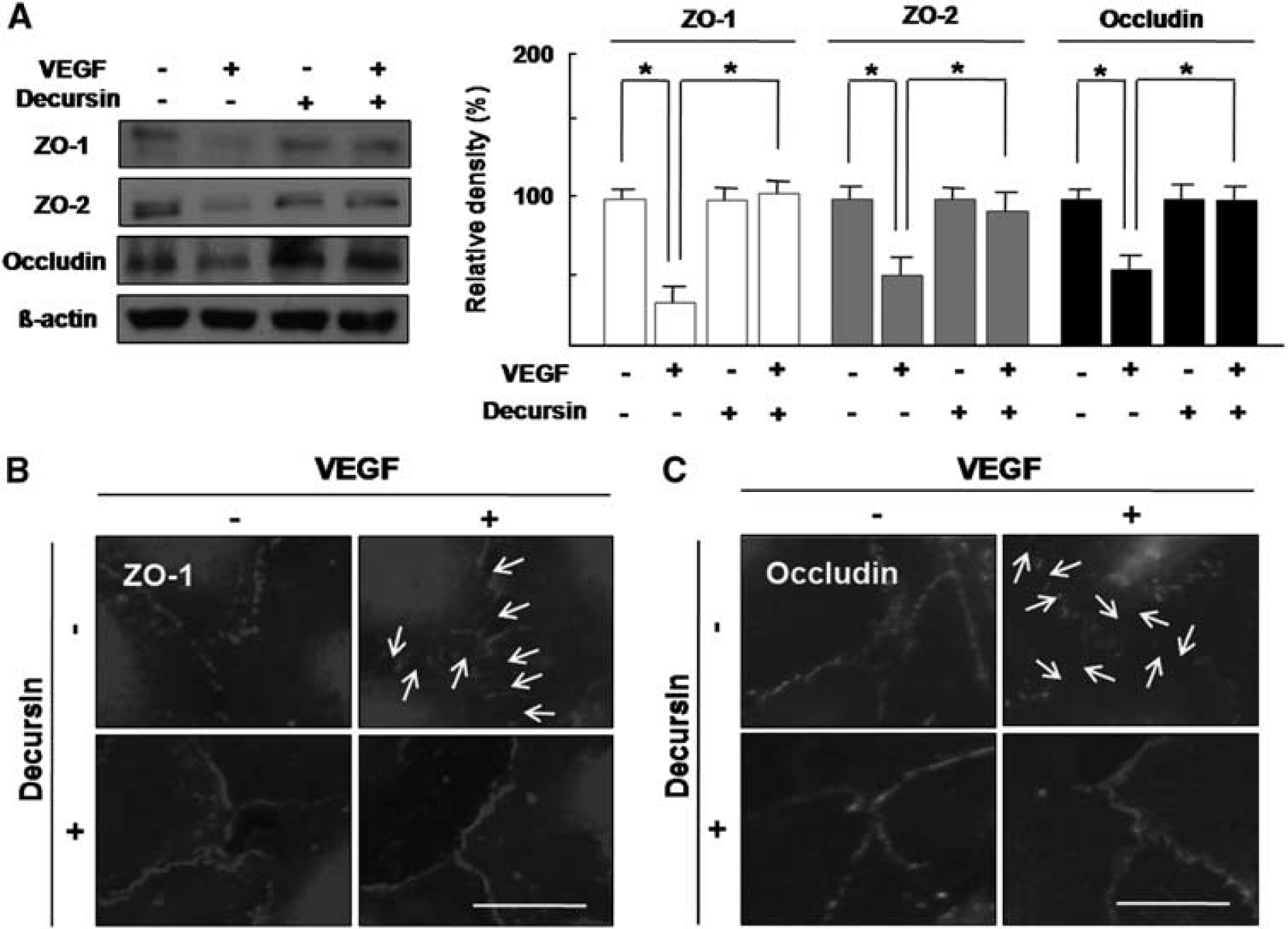
Decursin prevents VEGF-mediated loss of tight junction proteins in retinal endothelial cells. HRMECs were treated with 20 ng/mL VEGF or 10 μmol/L decursin. (
In addition, to confirm whether decursin prevents VEGF-mediated loss of tight junction proteins in intercellular junction of retinal endothelial cells, immunocytochemistry for ZO-1 and occludin was performed. In confluent HRMECs, ZO-1 and occludin contribute to the tight junction as cytoplasmic scaffolding protein and transmembrane protein, respectively (Kim et al, 2006). With treatment of 20 ng/mL VEGF, ZO-1 and occludin expression at intercellular junction were markedly reduced, which were completely recovered by cotreatment of 10 μmol/L decursin (Figures 2B and 2C).
Decursin Attenuates VEGF-Mediated Vascular Hyperpermeability in the Retina, Which Is Accompanied by Blockade of Loss of Tight Junction Proteins in Retinal Endothelial Cells
To investigate the antipermeable effect of decursin on VEGF-mediated vascular hyperpermeability, wholemount retinal preparation from 24 h after intravitreal injection of VEGF or decursin was performed after 1 h perfusion of fluorescein-conjugated dextran. As demonstrated in Figure 3A, decursin significantly inhibited diffuse leakage from retinal vessels in the VEGF-injected retina. With the intravitreal injection of 20 ng/mL VEGF, the fluorescein intensity outside retinal vessels increased 7-fold, whereas 10 μmol/L decursin significantly reduced fluorescein intensity to the level of control (P < 0.05).
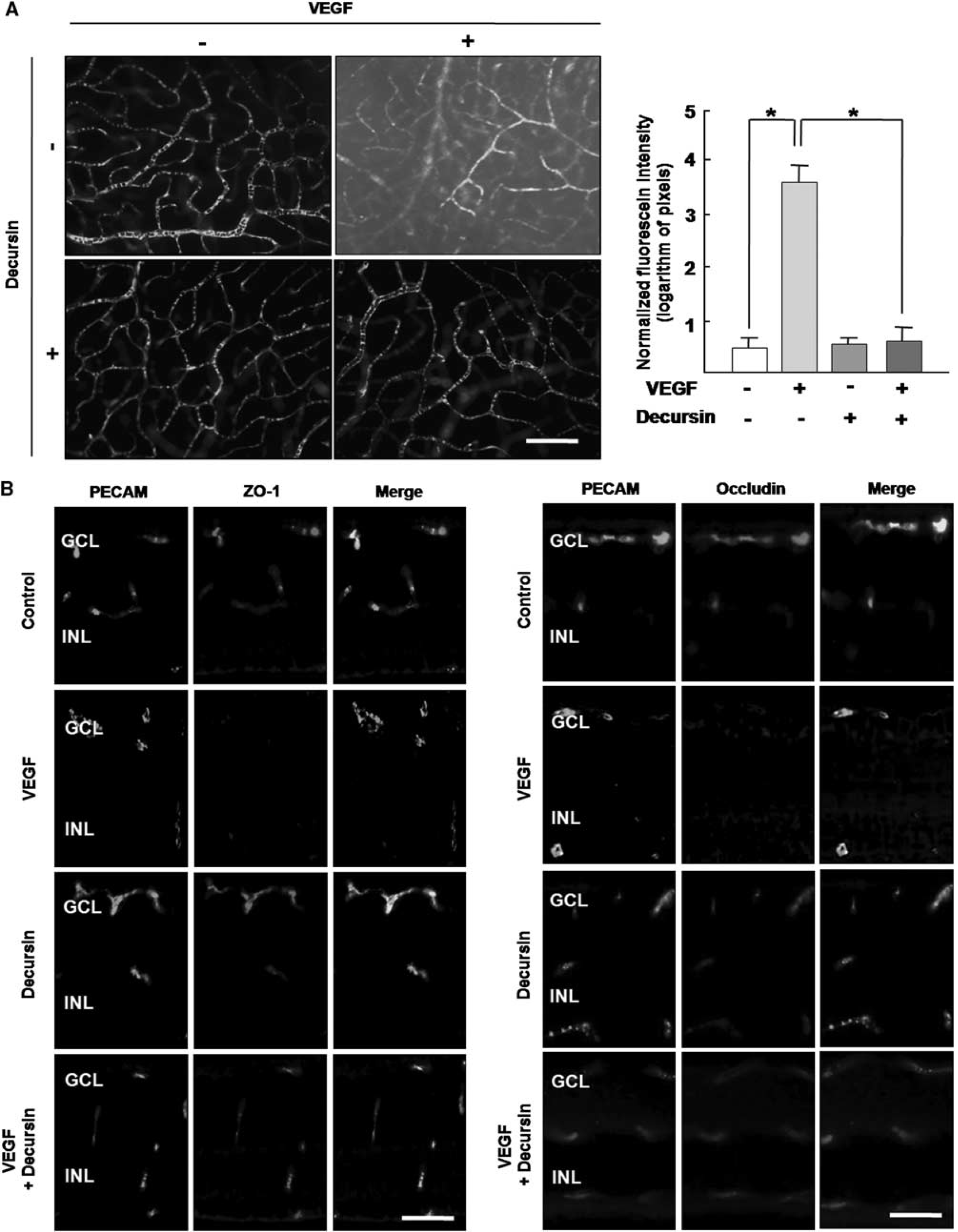
Decursin attenuates VEGF-mediated vascular hyperpermeability in the retina, which is accompanied by blockade of loss of tight junction proteins in retinal endothelial cells. (
To examine the preventive effect of decursin on VEGF-mediated loss of tight junction proteins in retinal vessels, immunohistochemistry for ZO-1 and occludin in the retina with the intravitreal injection of VEGF or decursin was performed. As shown in Figure 3B, ZO-1 and occludin in control were strongly expressed on retinal endothelial cells, which were well merged with PECAM, an endothelial cell marker. With 20 ng/mL VEGF injection, the expression of tight junction proteins was dramatically decreased, which were however completely prevented by the coinjection of 10 μmol/L decursin.
Decursin Inhibits VEGF-Induced VEGFR-2 Phosphorylation in Retinal Eodothelial Cells
On the basis of VEGFR-2, specific intracellular signaling cascades are essential for VEGF-mediated angiogenic response including proliferation, migration, cell survival, increased permeability in vascular endothelial cells (Ferrara and Davis-Smyth, 1997), to address whether decursin inhibits the VEGFR-2 signaling pathway in retinal endothelial cells, we investigated the inhibitory activity of decursin to VEGF-induced autophosphorylation of VEGFR-2 in HRMECs. An amount of 20 ng/mL VEGF significantly induced VEGFR-2 phosphorylation, which was however abrogated by cotreatment of 10 μmol/L decursin (P < 0.05, Figure 4A). With VEGF-mediated VEGFR-2 activation, ERK 1/2 was activated, but not Akt. VEGF-mediated activation of ERK 1/2 was also effectively inhibited by decursin treatment (P < 0.05, Figure 4B).
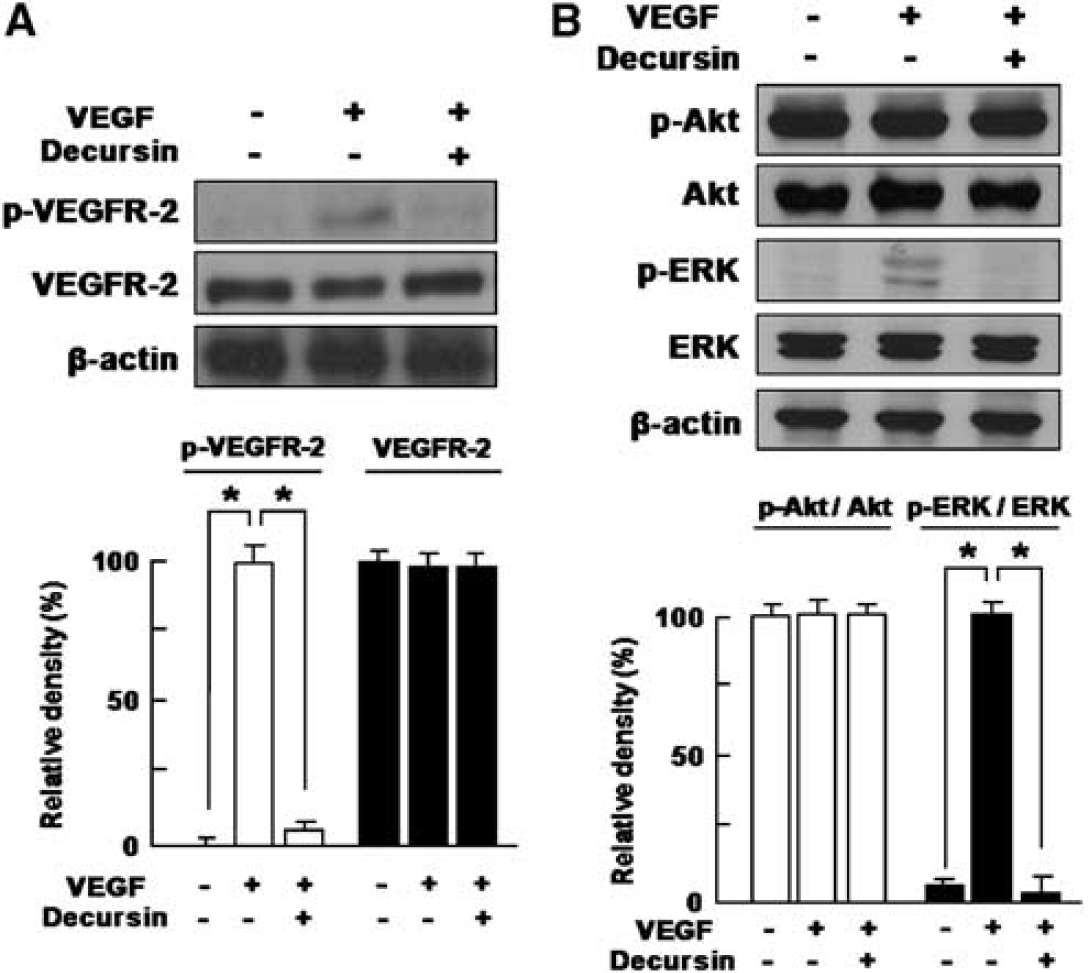
Decursin inhibits VEGF-induced VEGFR-2 phosphorylation in retinal eodothelial cells. HRMECs were treated with 20 ng/mL VEGF or 10 μmol/L decursin. Western blot analysis for (
Decursin Never Induces Cellular Toxicity in Retinal Endothelial Cells and Retinal Toxicity Under Therapeutic Concentrations
To investigate cytotoxicity of decrsin on retinal endothelial cells, MTT assay was performed in various concentrations of decursin (0.1 to 100 μmol/L). Up to 50 μmol/L decursin, five times of effective therapeutic concentration (Jung et al, 2009), decursin never affected the cellular viability of HRMECs (Figure 5A).
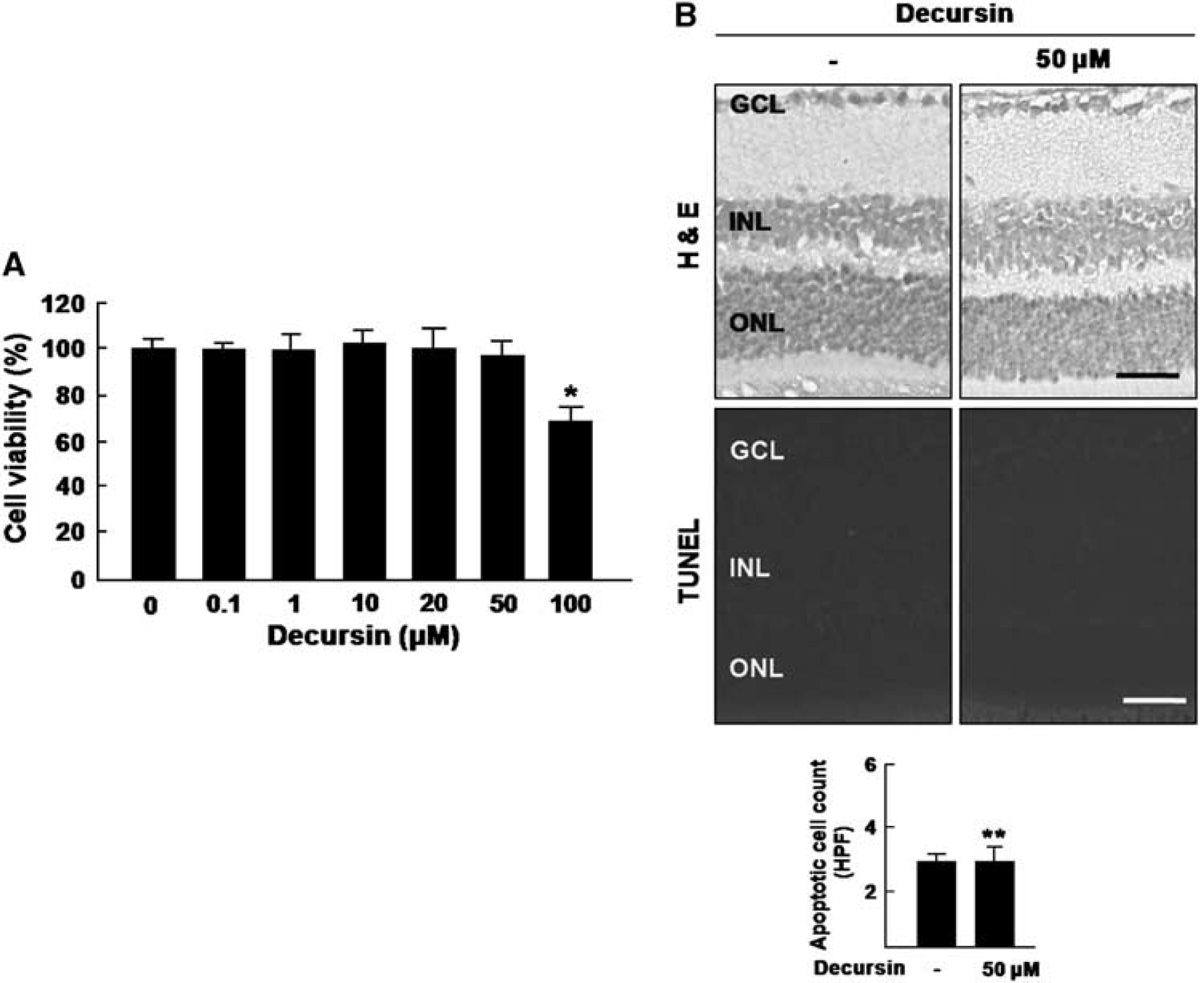
Decursin never induces cellular toxicity in retinal endothelial cells and retinal toxicity under therapeutic concentrations. (
In addition, to examine retinal toxicity of decursin, histologic examination and TUNEL assay were performed after intravitreal injection of 50 μmol/L decursin. As demonstrated in Figure 5B, even in high concentration of decusin, there was no change in retinal thickness and all retinal layers compared with control. Additionally, there was no inflammatory cell in the vitreous, retina, or choroid. Compared with control, TUNEL-positive cells were not increased with decursin injection (P > 0.05).
Discussion
In the course of our research for new angiogenesis inhibitors from natural products, we recently found out that decursin could be a potent angiogenesis inhibitor to suppress VEGF-mediated angiogenic processes in vitro and in vivo (Jung et al, 2009). Although transcellular as well as intercellular pathway could be altered in BRB breakdown, based on our earlier reports that VEGF-mediated alteration of tight junction proteins leads to BRB breakdown (Choi et al, 2007; Kim et al, 2009a), we focused on whether inhibition of VEGF-VEGFR-2 signaling pathway by decursin could block the alteration of intercellular tight junction and BRB breakdown. In this study, we clearly demonstrated that decursin inhibits VEGF-mediated loss of tight junction proteins in retinal endothelial cells and blocks VEGF-mediated vascular leakage in the retina, which is mediated by suppression of VEGFR-2 signaling pathway.
Hypoxic insult by retinal ischemia is an important cause of variable retinopathies to lead blindness. Hypoxia induces BRB breakdown, which is characterized by increased vascular permeability that is closely related to the alteration of tight junction proteins (Fischer et al, 1999; Kaur et al, 2008). The vascular permeability is defined as the movement of fluids and molecules across vascular endothelial cells caused by the disruption of intercellular endothelial junctions, which is mainly regulated VEGF-mediated signaling pathway in both physiologic and pathologic states (Weis and Cheresh, 2005; Kim et al, 2006). VEGF, originally known to be a vascular permeability factor, is one of the most well-known hypoxia-induced effectors, which is evidently mediated by the high-affinity cell surface receptors VEGFR-1 and VEGFR-2. Although VEGF could regulate the expression of tight junction proteins at the transcriptional and posttranslational levels, VEGF—VEGFR-2 signaling pathway is essential not only for angiogenesis processes including vascular endothelial proliferation, migration, and morphogenesis, but for vascular permeability (Ferrara and Davis-Smyth, 1997). As our previous reports, intercellular interactions to modulate both neural angiogenesis and tight junction formation play an important role in inner BRB formation (Lee et al, 2003b; Choi et al, 2007; Kim et al, 2009b). In addition, we have shown that the ischemia-induced or diabetic disruption of inner BRB might be due, in part, to alterations in the paracellular permeability pathway attributed to loosening of the tight junctions (Kim et al, 2007, 2009a), which suggests that well-organized tight junction molecules are requisite for the maintenance of barrier function in retinal vessel. In particular, ZO family and occludin are well-characterized components of the tight junction in retinal endothelial cells, whose expression are inversely related to permeability in BRB (Choi et al, 2007; Kim et al, 2007, 2009a,b). We herein showed that inhibition of VEGFR-2 activation by decursin is closely related to prevent VEGF-mediated loss of tight junction proteins including ZO-1/-2 and occludin to block BRB breakdown, which could be supported by our recent report that decursin inhibits VEGF-mediated in vitro and in vivo angiogenesis through suppression of VEGFR-2 activation (Jung et al, 2009). In addition, among diverse VEGFR-2-mediated signaling pathways including phosphatidylinositol-3-OH kinase/Akt, Ras/Raf/MEK/Erk, Src, and phospholipase C-γ/endothelial nitric oxide synthase (Takahashi et al, 1999, 2001), decursin effectively inhibited ERK 1/2 activation followed by VEGF-mediated VEGFR-2 phosphrylation.
Interestingly, decursin never affected cellular viability of retinal endothelial cells, and showed no retinal toxicity up to 50 μmol/L, five times of therapeutically effective concentration, which means that decursin could safely be applied to variable retinopathies of BRB breakdown without toxicity to retina and normal retinal vessels.
In summary, we demonstrated that decursin could inhibit VEGF-mediated inner BRB breakdown through suppression of VEGFR-2 signaling pathway. Decursin effectively and safely prevented VEGF-mediated hyperpermeability of retinal vascular endothelial cells and vascular leakage from retinal vessels, which were closely related to block loss of tight junction proteins. Interestingly, decursin suppressed VEGF-induced VEGFR-2 phosphorylation followed by the consequent inhibition of ERK 1/2 activation. Taken together, decursin, as a novel inhibitor to BRB breakdown, could be further developed for a therapeutic to prevent BRB breakdown in variable retinopathies.
Footnotes
The authors declare no conflict of interest.