
Abstract
There is a rising intracellular Zn2+ transient during neuronal ischemic hypoxia (oxygen-glucose deprivation and reoxygenation, OGD/R). The results of our recent works suggest that the OGD/R-induced Zn2+ transient can readily be mistaken for a Ca2+ transient. The aim of this study was to examine the respective functions of Zn2+ and Ca2+ in OGD/R-induced neuronal injury. We showed that [Zn2+]i accumulation was consistently met with the induction of OGD/R-induced cell injury. Ca2+ accumulation induced with high [K+] (to open voltage-gated calcium channels) or ionomycin (a Ca2+ ionophore) caused a moderate neuronal injury that was reduced significantly by the application of the Zn2+ chelator N,N,N',N'-tetrakis(2-pyridylmethyl)ethylenediamine (TPEN). In comparison, Zn2+ accumulation, induced with the Zn2+ ionophore pyrithione, resulted in significantly greater injury. The application of nimodipine and MK801 was shown to attenuate neuronal injury only from a mild (10 mins) OGD insult. Neuronal injury from more severe (30 mins) OGD was not mitigated by the ion channel antagonists, whereas treatment with the Zn2+ chelator TPEN did afford significant protection from cell injury. These results indicate Zn2+-mediated damage to be of greater consequence than Ca2+-mediated damage, and collectively support the suggestion that Zn2+ accumulation may be a more significant causal factor of OGD/R-induced neuronal injury.
Introduction
Neurons undergo changes in ion homeostasis as a result of normal physiologic stimuli. When these changes become uncontrolled, as in traumatic brain injury, ischemic stroke, and excitotoxicity, they cause pathologic dysfunction. Current models describing the causal events of excitotoxic brain injury focus on the concept of calcium (Ca2+) influx and accumulation as an immediate consequence of the massive release of glutamate seen during cerebral hypoxia/ischemia (Benveniste et al, 1988; Paschen, 1996; Perez Velazquez et al, 1997). The relationship between Ca2+ entry and neuronal death has been extensively studied to address the question of how excessive Ca2+ influx or Ca2+ overload might contribute to neuronal injury/death after ischemic insult or brain trauma. Accumulated evidence also suggests that Ca2+ may not be the only divalent metal cation involved in neural injury (Frederickson et al, 2005; Galasso and Dyck, 2007), and that Ca2+ detection may indeed be mitigated by the concomitant accumulation of zinc (Zn2+) in oxygen-glucose deprivation and reoxygenation (OGD/R; Stork and Li, 2006a).
The function of Ca2+ accumulation in ischemic hypoxia and oxidative stress has come under greater scrutiny recently (Stork and Li, 2006a). Although the rising [Ca2+]i has been conventionally detected with fluorescent Ca2+ indicators, it is recognized that the most commonly used Ca2+ indicators such as Fura-2 and Calcium Green-1 are not selective solely for Ca2+ but rather show a higher affinity to Zn2+ (Grynkiewicz et al, 1985; Martin et al, 2006; Thompson et al, 2002). Questions regarding the role of Ca2+ in these pathologies have become particularly relevant when one considers the widespread reports of rising Zn2+ during ischemia. Zn2+ at high concentration has been consistently shown to be a critical mediator of the neuronal death associated with experimental global ischemia (Koh et al, 1996; Lee et al, 2002; Sorensen et al, 1998; Tonder et al, 1990; Wei et al, 2004; Yin et al, 2002). We have recently suggested that the signal detected by fluorescent Ca2+ indicators can be primarily composed of a Zn2+ signal (Stork and Li, 2006a). An important note is that the putative cell death signaling pathway triggered by Zn2+ is strikingly similar to that reported to be triggered by Ca2+ (Galasso and Dyck, 2007; Zhang et al, 2007). In the present study we sought to evaluate the specific contributions made by Zn2+ and Ca2+ toward the development of OGD/R-induced injury. The OGD/R model has been widely used as an in vitro ischemic model to evaluate ischemic hypoxic brain injury. Although fluorescent detection may have historically misrepresented ischemic Ca2+ signals, large bodies of evidence support a Ca2+ transient as a critical contributor to ensuant injury/death. If this is the case, we are compelled to ask why there are two such similar signals, and which one (if not a combined effect of both) exerts a more fundamental or decisive function in neuronal injury/death. Our data suggest Zn2+ accumulation to be a greater causal factor for the development of neuronal death after hypoxic injury.
Materials and methods
Preparation of hippocampal slices and experimental procedures were implemented as described previously (Stork and Li, 2006a, b ). Hippocampal slices of 250 μm thickness were prepared from the brains of male Sprague—Dawley rats using a Vibratome (Series 3000 equipped with 900R refrigeration module; The Vibratome Company, St Louis, MO, USA). The artificial cerebrospinal fluid (ACSF) used during slice preparation and in experiments contained the following ion concentrations (in mmol/L): 121 NaCl, 1.75 KCl, 1.3 MgCl2, 1.25 KH2PO4, 26 NaHCO3, 2.5 CaCl2, and 10 glucose. All experiments were performed at room temperature unless otherwise noted. After preparation, slices were placed into custom-made holders and randomly separated into groups, then were put into 300 ml incubation chambers containing ACSF bubbled with 95% O2/5% CO2. All physiologic solutions were tested using a Vapro vapor pressure osmometer, where values of 295 to 305 mOsm were considered acceptable (Wescor, Logan, UT, USA).
In experimental treatment groups, all slices were subjected to the experimental treatments as indicated and then returned to normal ACSF for 2.5 h to allow time for the development of delayed cell injury/death. For hypoxic-ischemic stress, OGD treatments were administered by switching slices to identical chambers with ACSF lacking glucose (NaCl adjusted to 131 mmol/L) and reoxygenation (R) was implemented by return to normal ACSF. In each trial, slices in control groups were transferred to chambers containing normal ACSF for an equal time as OGD exposure of slices in treatment groups. To standardize any possible effects from slice manipulation during experiments, we also switched control slices between two identical ACSF-containing incubation chambers with the same time interval as in experimental groups.
After slices were subjected to their respective treatments and returned to normal ACSF for 2.5 h, they were placed in ACSF containing 5 μg/ml of the fluorescent viability indicator propidium iodide (PI) for 30 mins, rinsed 3 × in ACSF, and then fixed with 4% paraformaldehyde until fluorescence was measured the following day. In viable cells, PI is excluded from entering the lipid bilayer because of the molecule's size and charge, but in nonviable cells, the dye freely enters the damaged membrane to bind nucleic acids (DNA and RNA) and yield bright red fluorescence. Propidium iodide fluorescence was measured using a Zeiss Axiovert LSM 510 (confocal) microscope equipped with a x20 Plan-NeoFluar (0.8 NA) objective (Carl Zeiss Inc., Stuttgart, Germany). PI was excited using a 543 nm HeNe laser line and emission filtered using a 560 nm LP filter. Standardized z-projections of each slice were produced and fluorescent intensity was quantified using Zeiss LSM 510 software. A oneway analysis of variance was used to examine for significant differences between treatment groups where differences in PI fluorescence intensity between treatment groups were considered significant when P < 0.05. All data are expressed as the mean ± s.e.m. for measurements of PI fluorescent intensity and normalized to express cell injury/death measured from experimental treatment groups relative to that measured in control groups.
Colocalization experiments with PI and either Newport Green (NG), Calcium Green-1, or Fura-2 were performed with unfixed slices. In these experiments, slices were exposed to OGD and returned to normal ACSF for 90 mins unless otherwise noted. At the microscope, PI (5 μg/ml) and either NG, Calcium Green-1, or Fura-2 (all were used at a concentration of 10 μmol/L unless otherwise noted) were added to the slice perfusate and slices were given 5 mins to allow adequate dye saturation before imaging. Images used for colocalization analysis were obtained using an inverted Zeiss Axiovert LSM 510 confocal microscope. The colocalization of PI and NG/Calcium Green-1/Fura-2 staining was performed using sequential two-channel scans to avoid fluorescence cross talk. This technique yielded red and green images, which were then overlaid and analyzed using Zeiss software. As described previously (Stork and Li, 2006a, b ), all colocalization experiments were conducted with minimal laser intensity and with minimal exposure time, such that photobleaching was insignificant and no photobleaching corrections were made.
Chemicals and Reagents
The viability indicator PI, the L-type Ca2+-channel blocker nimodipine, the N-methyl-
Results
OGD/R Neuronal Injury is Coupled With Rising Zn2+ or Zn2+ Overload
In this study, the relationship between Zn2+ release and the development of neuronal injury in OGD/R was investigated in unfixed tissue by measuring the magnitude of colocalization between Zn2+ indicator fluorescence and PI fluorescence, a conventional marker for cell injury. Increased intracellular Zn2+ concentrations ([Zn2+]i) were detected with the fluorescent Zn2+ indicator NG, which responds to increases in [Zn2+]i with increased green fluorescence intensity. NG was carefully selected for the purposes of the present study. A leading advantage of NG use is its high selectivity for Zn2+ over other ions such as Ca2+ and Fe2+ (Haugland, 2005; Martin et al, 2006).
In this set of experiments, we examined the extent of pyramidal neuron damage resultant from OGD/R in hippocampal CA1 region. In slices that underwent OGD/R treatment, increased [Zn2+]i was consistently observed in CA1 pyramidal neurons as shown by significantly increased NG fluorescence (Figure 1B), supporting a robust increase in cytosolic labile Zn2+ after injury. We also used other fluorescent Zn2+ indicators (Zinpyr-1 and FluoZin-3) to confirm the increase in [Zn2+]i (data not shown; but see Stork and Li, 2006b). Co-staining with the fluorescent cell death marker PI revealed that increased NG labeling occurred specifically in Pi-positive, ischemically injured/dead pyramidal neurons (Figure 1A and 1B). Pearson's colocalization coefficient (r) for the PI and NG channels of each image showed r = 0.830 (P < 0.01). The r value was calculated from the average r measured from n = 14 pairs (red and green channel pairs) of 1,024 × 1,024 pixel images. The significant colocalization between NG and PI showed that rising [Zn2+]i was observed in significant coincidence with the of development of OGD/R-induced neuronal damage. When the Zn2+ chelator TPEN (10 μmol/L) was co-applied during OGD/R, there was significant reduction in NG- and PI-positive neurons compared with that measured for OGD/R without TPEN addition (Figure 1C).
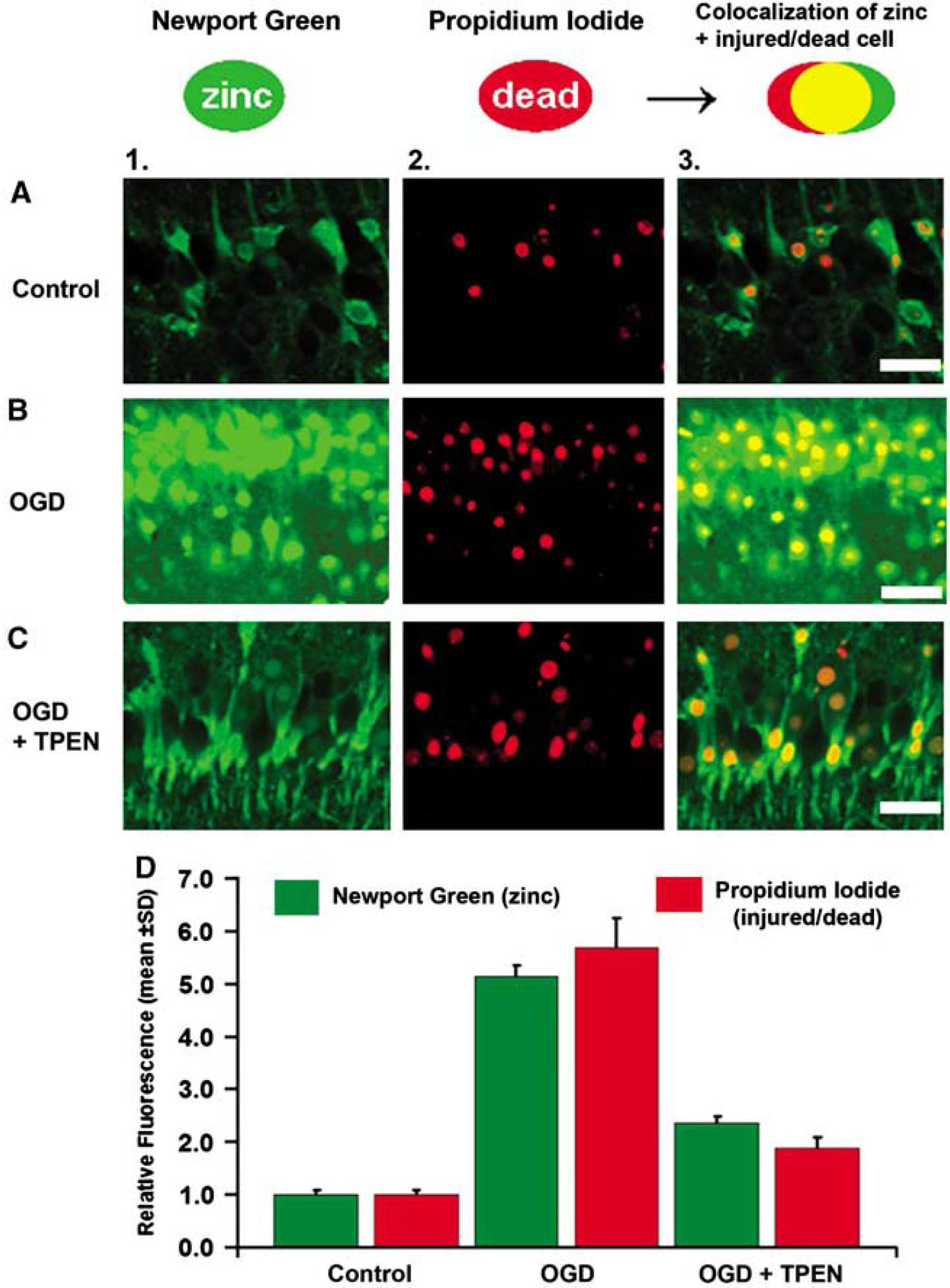
Colocalization study of rising zinc and cell death. Effects of ischemic conditions (OGD/R) on free Zn2+ levels and cell death shown in CA1 neurons by loading slices with the fluorescent Zn2+ indicator NG and the cell death indicator PI. Columns: 1, images of CA1 pyramidal neurons labeled with NG (10 μmol/L); 2, images of the same neurons labeled with PI; and 3, colocalized fluorescence of NG and PI. (
To verify that the observed increases of free Zn2+ seen in CA1 neurons exposed to OGD/R exerted direct neurotoxic effects, we conducted experiments to examine neuronal damage from induced [Zn2+]i increase. In this set of experiments, hippocampal slices were treated with exogenous Zn2+ (10 μmol/L) in combination with the Zn2+ ionophore Na-pyrithione (20 μmol/L) to induce an increase in [Zn2+]i. A Zn2+ concentration of 10 μmol/L was chosen because treatment with a 10 μmol/L concentration of TPEN was shown to have a significantly protective effect. It should be noted that the addition of 10 μmol/L Zn2+ (with pyrithione) does not necessarily equate to an intracellular concentration of 10 μmol/L. These treatments resulted in a marked increase of injured/dead neurons, shown by the significantly increased PI staining (Figure 4) as compared with controls. We chose to examine the toxicity of Zn2+ at a concentration of 10 μmol/L because we separately established that treatment with 10 μmol/L TPEN was significantly protective in OGD/R. Treatment of hippocampal slices with 10 μmol/L Zn2+ and 20 μmol/L pyrithione was shown to be highly toxic and results in significant injury (see Figure 4).
Colocalization of the Fluorescence of Calcium Indicators With PI
Currently available fluorescent Ca2+ indicators are sensitive to Zn2+. Figure 2 shows the colocalization of fluorescent Ca2+ indicators and PI in OGD/R injured CA1 neurons, done with unfixed slices. When hippocampal slices were stained with Calcium Green-1 and Fura-2, respectively, we saw increased fluorescence in CA1 pyramidal neurons of each OGD/R group. Figure 2A and Figure 2B show the high level of colocalization measured between Fura-2 and/or Calcium Green-1 with PI, with the colocalization coefficient r = 0.814 for Calcium Green-1 with PI (P < 0.01), and r = 0.827 for Fura-2 with PI (P < 0.01). Thus, rising Calcium Green-1 or Fura-2 fluorescence correlated with increased PI fluorescence after OGD/R. We further confirmed the ability of CaEDTA, a membrane impermeable Zn2+ chelator, to reduce ‘Ca2+’ fluorescence when we applied the chelator after OGD/R and after staining the slice with PI and Calcium Green-1 in the injured/dead neurons (Figure 2C). Note: TPEN (in Figure 1) was used before OGD or was co-applied during OGD to prevent neuronal injury. In OGD injured neurons that were stained with Calcium Green-1 and PI, CaEDTA selectively decreased Calcium Green-1 fluorescence, indicating the Zn2+ dependence of the signal.
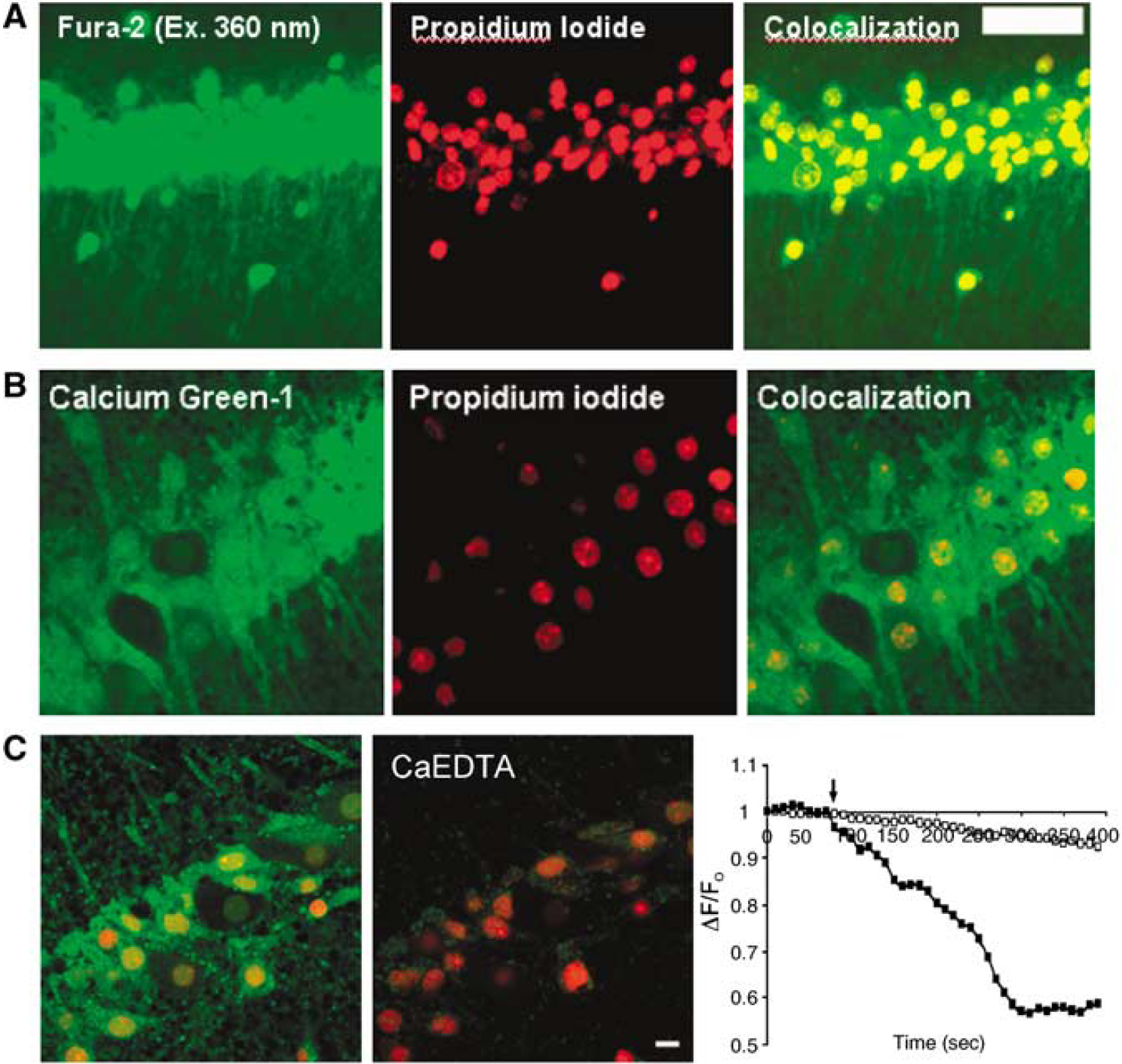
Colocalization of fluorescence Ca2+ indicators and PI in OGD. The fluorescence Ca2+ indicators Fura-2 (
Effect of Selectively Increased [Ca2+]i or [Zn2+]I on Neuronal Damage
In this study, excessive Ca2+ influx was induced by membrane depolarization using KC1 and separately, by the application of the Ca2+ ionophore, ionomycin. High concentrations of KC1 have been used to depolarize neuronal membrane potential and result in the opening of voltage-gated calcium channels, leading to Ca2+ influx propelled by its concentration gradient. Ionomycin increases plasma membrane permeability to Ca2+ and results in the shuttling of Ca2+ across the plasma membrane.
In this set of experiments, slices were exposed to ACSF containing 90 mmol/L KC1 and 5 mmol/L CaCl2 (NaCl adjusted to 25.25 mmol/L) for 10 mins then returned to normal ACSF for 2.5 h. To make the most of the experimental manipulation, we applied a higher concentration of Ca2+ to achieve a large increase in [Ca2+]i. In separate experiments, slices were exposed to ACSF containing 2.5 μmol/L of the Ca2+ ionophore ionomycin and 5 mmol/L CaCl2 (NaCl adjusted to 118.5 mmol/L) for 30 mins and then returned to normal ACSF for 2.5 h before PI staining and fixation. As expected, increased cell injury was observed after each treatment. The fluorescence data were normalized and compared to the control (without treatment) (Figure 3). Thus, these results indicate that increases in [Ca2+]i facilitate neuron injury, and support the current view that a Ca2+ transient is involved in ischemic cell death. To test for the involvement of an injurious Zn2+ transient after induced Ca2+ elevation, we applied a low concentration of Zn2+ chelator(s) in some trials. Notably, cell injury/death after the application of high KC1 or ionomycin was significantly prevented when the Zn2+ chelator TPEN (10 μmol/L) was co-applied (with high concentration K+ or ionomycin) (Figure 3). These particular data suggest that altered [Zn2+]i may mediate cell damage after excessive Ca2+ influx.
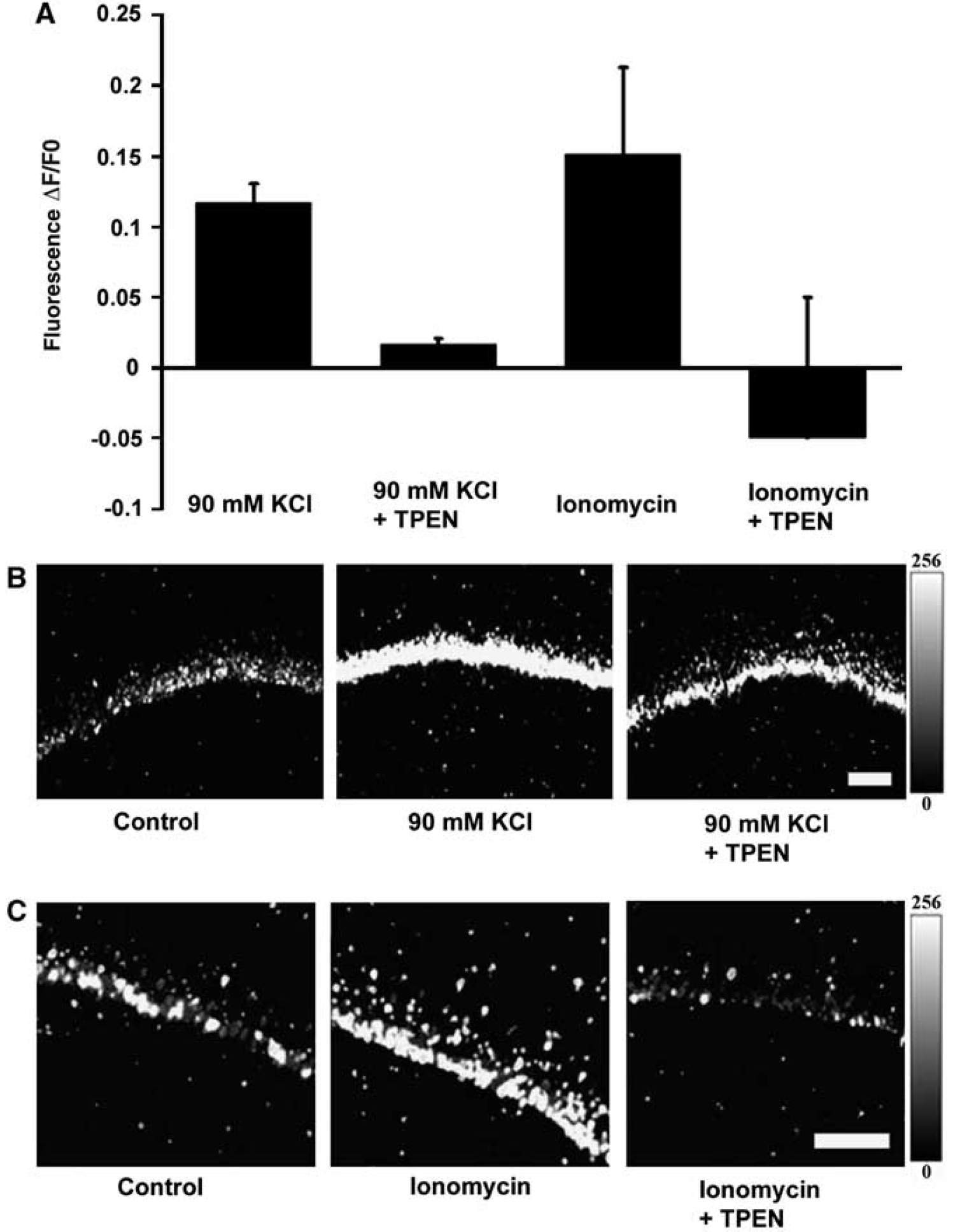
Neuronal damage induced by the influx of Ca2+ and its sensitivity to TPEN. Protective effects of the Zn2+ chelator TPEN from neuronal injury induced by treatments designed to elevate intracellular [Ca2+]. (
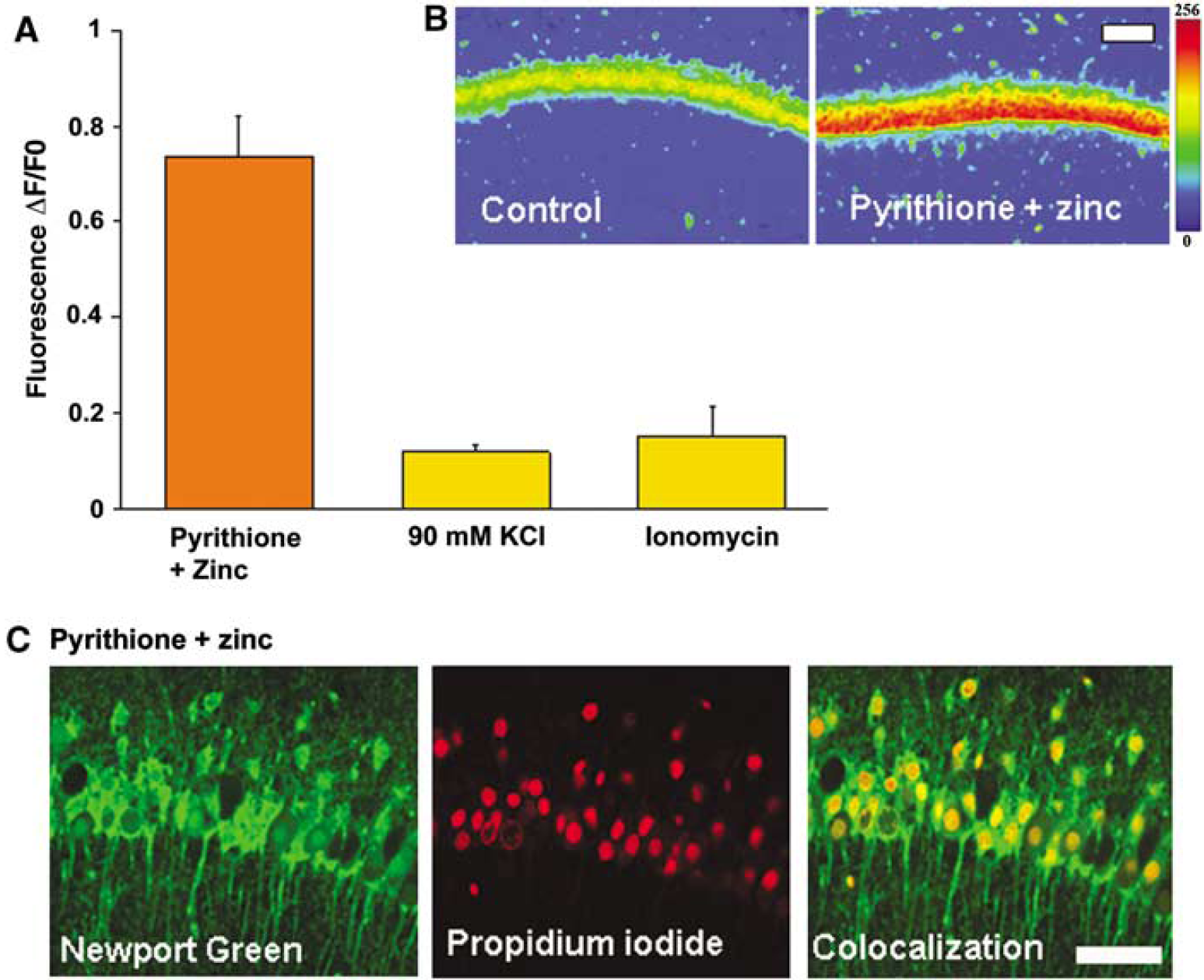
Neuronal injury induced by the influx of Zn2+. Injurious effect of Zn2+ pyrithione (to elevate intracellular Zn2+) on CA1 neurons in acute rat hippocampal slices. (
A key observation from the following trials was that the cell damage induced by Ca2+ influx was significantly less compared to the damage induced by Zn2+ influx (Figure 4A and 4B). The influx of Zn2+ was facilitated by the Zn2+ ionophore, Na-pyrithione. The measured PI fluorescence showed that the application of the same Zn2+ ionophore (20 μmol/L) with 10 μmol/L Zn2+ increased intracellular Zn2+ and effected significant cell injury/death (Figure 4C). It is worthwhile to note that a comparatively low concentration of exogenous Zn2+ was applied in these tests (contrasted with the 2 to 5 mmol/L application of exogenous Ca2+; see Figure 3), yet this Zn2+ treatment was seen to produce the greatest magnitude of cell death compared to that measured after all other injurious treatments in the present study.
Effect of Reducing Ca2+ Influx on OGD/R-Induced Neuronal Injury
Here we used two widely used agents, the NMDA receptor antagonist MK801 (10 μmol/L) and the L-type voltage-dependent calcium channel antagonist nimodipine (10 μmol/L). The relative protective effect of antagonist treatment was compared with the effect of Zn2+ chelation by applying each treatment during OGD. Cell viability was assessed by PI staining after the reoxygenation period in these trials. Fluorescent viability data from the antagonist treatment were normalized and compared to the control (OGD/R without treatment) and OGD/R in the presence of TPEN (10 μmol/L). A protective effect from MK801 and nimodipine was observed for a 10 mins OGD insult (Figure 5B), with protection absent for a 30 mins OGD insult (Figure 5A). Thus, this strategy of reducing Ca2+ influx gave protection during a shorter OGD duration, but showed no protective effect for a longer (30 mins) OGD insult. Application of TPEN in OGD significantly reduced neuronal death for a 10 or 30 mins OGD insult.
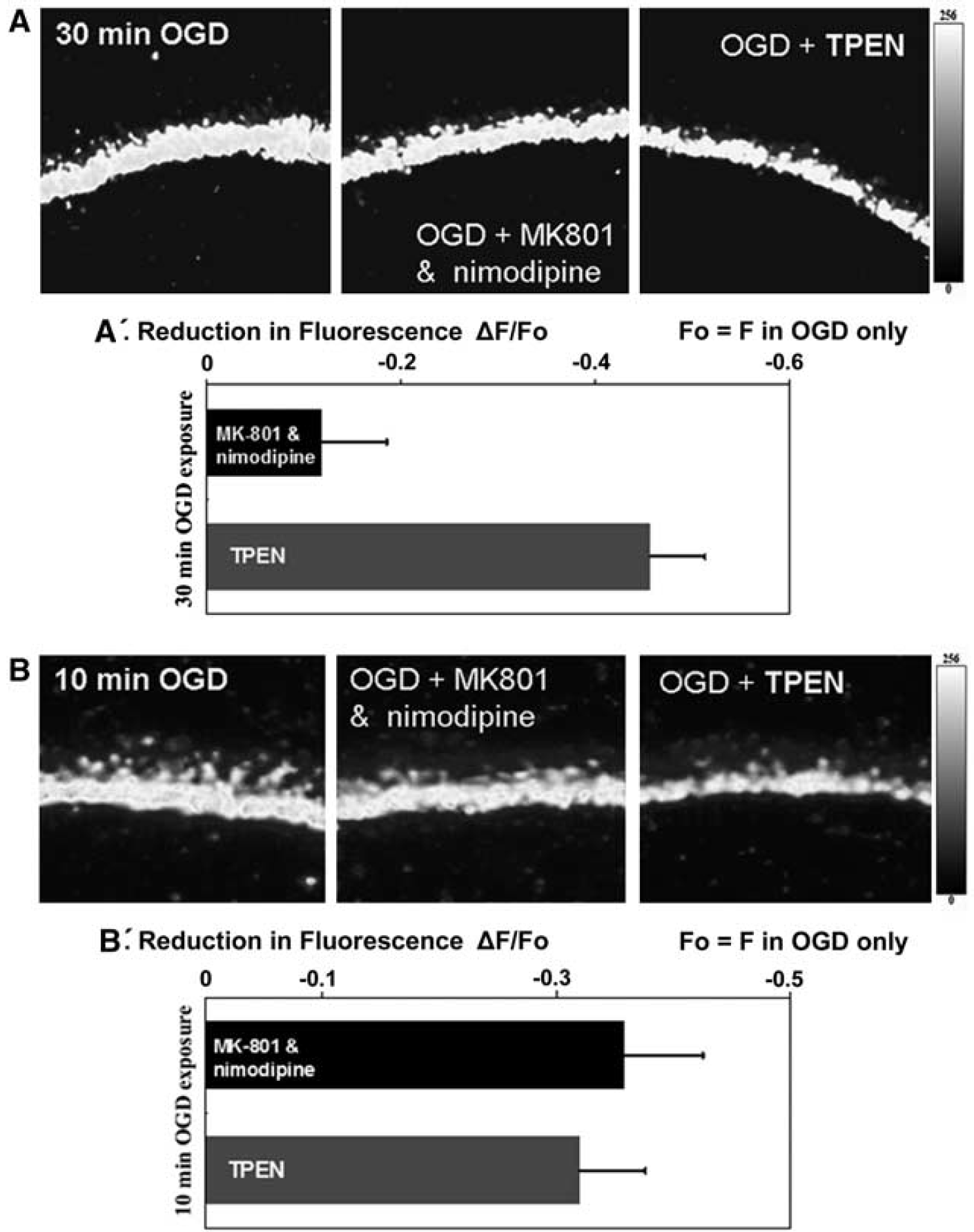
Effect of Ca2+ channel blockers on 10/30 min OGD. Relative protective effects of nimodipine, MK801, and TPEN on CA1 pyramidal neurons. PI fluorescence was recorded from the CA1 region of hippocampal slices given the indicated treatments. (
Discussion
The major findings are: (1) the increase in [Zn2+]i induced by OGD/R occurs coincident with neuronal injury/death in CA1 hippocampal pyramidal neurons; (2) the neuronal damage resultant from the induced Ca2+ rise was to be largely Zn2+ dependent, because the damage was significantly reduced by the application of TPEN; (3) further, these data show that Zn2+ elevation exerted potent neurotoxicity, resulting in more neuronal injury than the injury by the influx of extracellular Ca2+; (4) ion channel antagonism by nimodipine and MK801 gave a protective effect only for short (10 mins) OGD exposure, whereas TPEN showed significant protection for the longer (30 mins) exposure of OGD.
Zinc is a Causal Factor in OGD/R-Induced Neuronal Injury
The level of labile or free Zn2+ ions in the cytosol is normally low (~1 pmmol/L) because of tight regulation of Zn2+ homeostasis by cellular mechanisms (Frederickson and Bush, 2001; Outten and O'Halloran, 2001). As such, in cells with functioning Zn2+ regulatory mechanisms, the zinc ions are sequestered within proteins, as structural or catalytic cofactors, and thereby unavailable to coordinate with fluorescent indicators. In this way, the very low level of free Zn2+ found in healthy cells makes them appear ‘invisible’, to detection with fluorescent Zn2+ indicators. When [Zn2+]i increases beyond a normal range, such that the cell would become visible (detected with Zn2+ fluorescence indicators) after OGD, the increased cytosolic Zn2+ equates to an undesirable event and is certain to exert detrimental effects on neurons in OGD/R conditions. This speculation is supported by three observations: (1) reduction of [Zn2+]i in OGD/R with TPEN significantly reduces CA1 neuronal damage, supporting [Zn2+]i elevation as a damaging event in neurons; (2) the release of endogenous intraneuronal Zn2+ is consistently detected after cells underwent OGD/R, showing that elevated free Zn2+ was highly predictive of subsequent cell injury/death; (3) induced [Zn2+]i elevation was shown to be potently toxic. Further, the injury caused by increased Zn2+ was of greater severity than that caused by the influx of Ca2+.
Recent work by Vander Jagt et al (2008) has highlighted a potential interplay between Ca2+ and Zn2+ signals. Using a hippocampal slice model, it was shown that sustained NMDA exposure resulted in an initial intracellular increase of both Ca2+ and Zn2+ followed by a delayed progression of neurons into lethal Ca2+ overload (Vander Jagt et al, 2008). Intracellular Zn2+ chelation with TPEN delayed the progression of Ca2+ overload after NMDA exposure (Vander Jagt et al, 2008), pointing out a potential dualistic synergism of the ions' effects.
Sources of Intracellular Zn2+ Elevation
Growing evidence suggests that Zn2+ can be liberated from intracellular stores after oxidative stress and that accumulation of cytoplasmic Zn2+ is linked into a cascade of events leading to neuronal death (Bossy-Wetzel et al, 2004; Cuajungco and Lees, 1998; Land and Aizenman, 2005; Malaiyandi et al, 2004; Stork and Li, 2006b; Zhang et al, 2004). In viable CA1 neurons, Zn2+ is normally tightly bound to cellular proteins and enzymes and apparently meticulously regulated, limiting the extent of detectable intracellular free Zn2+ concentrations in eukaryotic cells. The CA1 area of the hippocampus was chosen as the region of interest because of its increased vulnerability to ischemic damage and because the results from CA1 analysis can be readily interpreted within the context of the extensive literature regarding this hippocampal region. The lack of detectable free Zn2+ found in CA1 neurons is consistent with a view of Zn2+ homeostasis being meticulously regulated in normal neurons, where the concentrations of free [Zn2+]i are generally considered to be in the low picomolar to low nanomolar range (Krezel and Maret, 2006; Outten and O'Halloran, 2001). Our results (also see Wei et al, 2004) indicate substantial amounts of Zn2+ accumulation occurs in OGD/R, as shown by detection with the low affinity indicator NG (KD zinc = 1 to 3 μmol/L). Whether other means of Zn2+ storage exist, alike mechanisms for Ca2+ storage, is unknown. One proposed buffering system is that performed by metallothioneins, which are amplified in number in response to Zn2+ elevation, and thus provide strong buffering capacity for cytosolic Zn2+ (Maret, 1994). A protective function of the Zn2+ -binding protein metallothionein in ischemic cell death has been proposed (van Lookeren Campagne et al, 1999).
Zn2+ Contributes to ‘Ca2+ Overload’
The influx of Ca2+ and the subsequent triggering of Ca2+ release from intracellular stores are generally believed to mediate the rapid cellular demise seen in ischemia. Although we recognize that many published studies support the function of Ca2+, or Ca2+ overload, in ischemicstroke; a majority of these studies have relied on fluorescent Ca2+ indicators to detect Ca2+. Because available fluorescent Ca2+ indicators show a greater sensitivity to Zn2+ and because nearly all Ca2+ chelators tend to chelate Zn2+ with greater affinity, difficulty remains in the understanding of the action of Ca2+ itself (Martin et al, 2006; Stork and Li, 2006a). This currently becomes much less reliable for evaluation solely of the intracellular Ca2+ transient's effect and even more difficult to evaluate specifically its effect on cell damage independently from Zn2+. Notwithstanding, we assert that these studies by no means diminish the importance of Ca2+ signaling; and in fact, our observations are intended to ultimately facilitate a better understanding of the function of Ca2+ in ischemic hypoxic brain injury. It is our opinion that an accord on the specific effects of Ca2+ signaling in ischemic hypoxic injury will remain out of reach until the advent of truly specific Ca2+ chelators; allowing for the singular manipulation of this ion during excitotoxic or hypoxic condition.
Induced Zn2+ Elevation Shows Greater Toxicity Than Induced Ca2+ Elevation
At the center of the Ca2+ overload hypothesis is the notion that the influx of extracellular Ca2+ triggers a rise in [Ca2+]i subsequently resulting in cellular injury/death. However, in two scenarios designed to induce Ca2+ influx (KC1 and ionomycin treatment), results showed significantly less injury, as compared to that by Zn2+ influx induced with pyrithione (Figure 4). Interestingly, the neuronal injury initiated by Ca2+ influx was largely prevented by Zn2+ chelation (Figure 3), suggesting a link between Ca2+ influx and Zn2+ dyshomeostasis. This may be explained by the effects of Ca2+ influx in triggering the release of Zn2+ from its intracellular storage sites. One recent investigation that also used acute hippocampal slices in an OGD/R model of ischemia observed an initial Zn2+ transient that preceded and accelerated the development of Ca2+ deregulation (Medvedeva et al, 2009). The investigators showed that Zn2+ chelation with TPEN during OGD/R delayed the onset of Ca2+-mediated destruction of the plasma membrane (Medvedeva et al, 2009). The present work, in addition to recently published results, collectively suggest the interplay of OGD/R-induced Ca2+ and Zn2+ transients leading to neuronal demise. Ongoing work in our lab also indicates a fundamental interplay of Ca2+ and Zn2+ signals, wherein we have observed that Zn2+ can be released from thapsigargin-sensitive stores (unpub-lished data). One likely point of convergence for these ion signals has been suggested by recent work showing Ca2+ to initiate nitric oxide synthase activation and NO production leading to intracellular Zn2+ release (Bossy-Wetzel et al, 2004). Another point of interaction is likely to be the mitochondria. Several studies suggest that Zn2+ induces neuronal death by injury to the mitochondria (Bonanni et al, 2006; Bossy-Wetzel et al, 2004; Dineley et al, 2003; Gazaryan et al, 2007; Jiang et al, 2001).