
Abstract
Babies experience hypoxia (H) and ischemia (I) from stroke. The only approved treatment for stroke is fibrinolytic therapy with tissue-type plasminogen activator (tPA). However, tPA potentiates H/I-induced impairment of responses to cerebrovasodilators such as hypercapnia and hypotension, and blockade of tPA-mediated vasoactivity prevents this deleterious effect. Coupling of tPA to red blood cells (RBCs) reduces its central nervous system (CNS) toxicity through spatially confining the drug to the vasculature. Mitogen-activated protein kinase (MAPK), a family of at least three kinases, is upregulated after H/I. In this study we determined whether RBC-tPA given before or after cerebral H/I would preserve responses to cerebrovasodilators and prevent neuronal injury mediated through the extracellular signal-related kinase (ERK) MAPK pathway. Animals given RBC-tPA maintained responses to cerebrovasodilators at levels equivalent to pre-H/I values. cerebrospinal fluid and brain parenchymal ERK MAPK was elevated by H/I and this upregulation was potentiated by tPA, but blunted by RBC-tPA. U0126, an ERK MAPK antagonist, also maintained cerebrovasodilation post H/I. Neuronal degeneration in CA1 hippocampus after H/I was not improved by tPA, but was ameliorated by RBC-tPA and U0126. These data suggest that coupling of tPA to RBCs offers a novel approach toward increasing the benefit/risk ratio of thrombolytic therapy for CNS disorders associated with H/I.
Introduction
Ischemic stroke is an important contributor to morbidity and mortality. Pediatric stroke can occur in as many as 1 in 4,000 births (Nelson and Lynch, 2004) and complications because of hypoxia/ischemia (H/I) are common (Ferriero, 2004). Maternal and perinatal coagulopathy predispose to pediatric stroke (Gunther et al, 2000; Kraus and Acheen, 1999) with 30% of such events occurring because of thrombosis (DeVeber and Andrew, 2001). The thrombolytic agent tissue-type plasminogen activator (tPA) remains the only approved treatment for acute stroke (Kim et al, 1999), but its use in children has been limited and its benefit remains unclear (Benedict et al, 2007; Janjua et al, 2007). Indeed, the brief therapeutic window of tPA and the high incidence of posttreatment complications, including intracranial hemorrhage (ICH), have constrained the actual clinical use of tPA to approximately 3% to 8% of all patients eligible for such therapy (Lapchak and Araujo, 2001; Lapchak, 2002). Patients often remain hospitalized after an initial ischemic event because of the risk of recurrent cerebrovascular thrombosis, indicating a clear unmet need for safer and more effective treatment and thromboprophylaxis.
Balancing the risks and benefits of tPA is one of the central issues in managing patients with stroke and other cerebrovascular disorders accompanied by thrombosis and ischemia. Although the beneficial effects of tPA reducing the clot burden are apparent, the drug may also increase the volume of injured tissue after stroke, as exemplified in tPA null mice, provoke ICH, and exacerbate excitotoxic neuronal death by enhancing signaling through the
Recent studies of brain ischemia implicate signaling through the low-density lipoprotein (LRP)-receptor in the pathologic effects of plasminogen activators on the cerebrovasculature (Armstead et al, 2008). Our group has shown that a plasminogen activator inhibitor-1-derived peptide (EEIIMD) inhibits tPA- and LRP-mediated signal transduction without compromising catalytic activity (Armstead et al, 2005a; Akkawi et al, 2006; Nassar et al, 2004). Specifically, EEIIMD blocks the deleterious effects of exogenous tPA on stroke volume, ICH, and edema in models of rat middle cerebral artery occlusion and embolic stroke models and markedly prevents neuronal cell loss after traumatic brain injury (TBI) in piglets without inhibiting the fibrinolytic activity of tPA(Armstead et al, 2006). These data indicate that it may be possible to improve the clinical outcome of a stroke and TBI by separating the favorable and harmful activities of tPA.
In animal models of stroke, tPA contributes to an adverse outcome through mechanism(s) involving the activation of matrix metalloproteinases (MMPs) (Tsuji et al, 2005). Matrix metalloproteinases are upregulated after brain injury (Wang et al, 2002; Laher and Zhang, 2001), in part, by activating mitogen-activated protein kinase (MAPK), a family of at least three kinases (extracellular signal-related kinase—ERK, p38, and c-Jun-N-terminal kinase—). Our recent studies show that the urokinase plasminogen activator contributes to impaired stimulus-induced cerebrovascular dilation after cerebral H/I in the newborn pig through LRP and upregulation of ERK MAPK (Armstead et al, 2008).
Contemporaneous studies from our group have shown that anchoring tPA on red blood cells (RBC) endows the resultant complex, RBC-tPA, with dramatically prolonged circulation time (many hours versus minutes for tPA), while spatially constraining it to the intravascular space (Murciano et al, 2003; Ganguly et al, 2005; Zaitsev et al, 2006). In rodent models of cerebrovascular thrombosis and TBI, treatment with this RBC-tPA complex provided effective thromboprophylaxis, rapid reperfusion, neuroprotection, and reduction in mortality all without causing ICH (Danielyan et al, 2008; Stein
This study was designed to determine whether RBC-tPA given before or after cerebral H/I would help preserve stimulus-induced cerebrovasodilation and prevent neuronal injury mediated through the ERK MAPK pathway.
Materials and methods
Closed Cranial Window Technique and Cerebral Hypoxia/Ischemia
Newborn pigs (1 to 5 days; 1.2 to 1.8 kg) of either sex were studied. All protocols were approved by the Institutional Animal Care and Use Committee. Animals were sedated with isoflurane (1 to 2 MAC). Anesthesia was maintained with a-chloralose (30 to 50 mg/kg, supplemented with 5 mg/kg/h i.v.). A catheter was inserted into a femoral artery to monitor blood pressure and to sample for blood gas tensions and pH. Drugs to maintain anesthesia were administered through a second catheter placed in a femoral vein. The trachea was cannulated, and the animals were ventilated with room air. A heating pad was used to maintain the animals at 37°C to 39°C, monitored rectally.
A cranial window was placed in the parietal skull of these anesthetized animals. This window consisted of three parts: a stainless steel ring, a circular glass coverslip, and three ports consisting of 17-gauge hypodermic needles attached to three precut holes in the stainless steel ring. For placement, the dura was cut and retracted over the cut bone edge. The cranial window was placed in the opening and cemented in place with dental acrylic. The volume under the window was filled with a solution, similar to cerebrospinal fluid (CSF), of the following composition (in mmol/L): 3.0 KC1, 1.5 MgCl2, 1.5 CaCl2, 132 NaCl, 6.6 urea, 3.7 dextrose, and 24.6 NaHCO3. This artificial CSF was warmed to 37°C and had the following chemistry: pH 7.33, pCO2 46 mmHg, and pO2 43 mmHg, which was similar to that of endogenous CSF. Pial arterial vessel diameter was measured with a microscope, a camera, a video output screen, and a video microscaler.
Total cerebral ischemia was accomplished by infusing artificial CSF into a hollow bolt in the cranium to maintain an intracranial pressure 15 mmHg greater than the numerical mean of systolic and diastolic arterial blood pressure. Intracranial pressure was monitored via a sidearm of the cranial window. To prevent the arterial pressure from rising inordinately (Cushing response), venous blood was withdrawn as it was necessary to maintain mean arterial blood pressure no greater than 100 mmHg. As the cerebral ischemic response subsided, the shed blood was returned to the animal. Cerebral ischemia was maintained for 20 mins. Hypoxia (pO2 of approximately 35 mmHg) was produced for 10 mins before ischemia by decreasing the inspired O2 via inhalation of N2, which was followed immediately by total cerebral ischemia. Hypotension was induced by the rapid withdrawal of either 5 to 8 or 10 to 15 mL blood/kg to induce moderate or severe hypotension (decreases in mean arterial blood pressure of 25% and 45%, respectively). Such drops in blood pressure were maintained constant for 10 mins by titration of additional blood withdrawal or blood reinfusion. Two levels of hypercapnia (low and high) were induced via inhalation of the graded levels of a 10% CO2 -to 21% O2 to balance the N2 gas mixture for 10 mins to produce levels of pCO2 of 50 to 60 mmHg for low exposure and 70 to 80 mmHg for high exposure.
Protocol
Two types of pial vessels, small arteries (resting diameter, 120 to 160 μm) and arterioles (resting diameter, 50 to 70 μm), were examined to determine whether segmental differences in the effects of H/I could be identified. Typically, 2 to 3 mL of artificial CSF were flushed through the window over a 30-secs period, and excess CSF was allowed to run off through one of the needle ports. For sample collection, 300 μL of the total cranial window volume of 500 μL was collected by slowly infusing artificial CSF into one side of the window and allowing the CSF to drip freely into a collection tube on the opposite side.
Thirteen experimental groups were studied (all
Preparation of RBC-tPA
Red blood cells were isolated by centrifugation from fresh anticoagulated (heparin, 1,000 U/kg) animal blood. Biotinylated tPA was coupled with biotinylated RBC via streptavidin, producing RBC-tPA complexes possessing 5 × 104 tPA molecules per RBC, as described earlier (Murciano et al, 2003; Ganguly et al, 2005).
Enzyme-Linked Immunosorbent Assay
Commercially available enzyme-linked immunosorbent assay (ELISA) kits were used to quantity the CSF ERK MAPK (Assay Designs, Ann Arbor, MI, USA) concentration. Phosphorylated ERK MAPK enzyme values were normalized to total form and then expressed as a percentage of the control condition.
Immunohistochemistry
Four hours after cerebral H/I, the animal was killed and two thin slices of parietal cortex were cut parallel to the brain surface. These slices were placed in 4% paraformaldehyde for 24 h at 4°C and then subjected to paraffin sectioning. Paraffin sections of the parietal cortex from piglet brains after H/I and from uninjured sham control animals were unwaxed, were incubated in 10 mmol/L sodium citrate buffer pH 6.0 inside a food steamer (subboiling temperature) for 10 mins to unmask the antigen, endogenous peroxidase was blocked with 0.3% H2O2, and stained with anti-phospho-p44/42 MAPK rabbit monoclonal antibody, which recognized the phosphorylated forms of both p42 and p44 kinases (ERK1 and ERK2) (1 μg/mL, Cell Signaling #4376, Billerica, MA, USA), or with mouse IgG1 as a negative control, secondary biotinylated antimouse IgG (1:200), followed by incubation with horseradish peroxidase- (HRP)-conjugated streptavidin. Peroxidase was detected using the avidin—biotin complex ABC kit (Vector Lab, Burlingame, CA, USA) counterstained with hematoxylin. Positive staining is visualized by the brown-colored [3,3-diaminobenzidine] reaction product.
Histologic Preparation
The brains were perfused with heparinized saline, followed by 4% paraformaldehyde and then phosphate-buffered saline. Coronal sections were prepared at 0.5 cm intervals for gross examination and photography. For histopathology, staining was performed on both frozen and paraffin-embedded slides, blocks from some animals were cryoprotected in sucrose, and serial sections were cut at 40 μm intervals from the front face of each frozen block and mounted on microscope slides. The sections (10 μm) were stained with hematoxylin and eosin (HE). Six animals from each of the groups were studied.
Histologic Analysis of Degeneration of Neurons
All procedures were performed by investigators who were blinded to treatment. Preliminary examinations of injured brains revealed that the parietal lobe and the hippocampus suffered the most extensive pathologic changes. Such more detailed histopathologic analyses were performed in these regions. The specific anatomic location used to select sections was based on the Stereotaxic Atlas of the Pig Brain (Felix et al, 1999). To determine the extent of neuron degeneration in the cortex, six HE-stained sections from both hemispheres from each animal were examined and the number of dying neurons was counted manually. Fifteen frames from each slide (1 × 1.2 mm per frame) encompassing a total area of 18 mm2 were chosen to estimate the number of degenerating neurons in the parietal cortex using light microscopy at × 100 magnification. To determine the extent of neuron degeneration in the hippocampus, six HE-stained sections from both the hemispheres from each animal were examined and the number of dying neurons in the CA1 subfield was counted manually. This subfield has been found earlier to display the most extensive neuronal degeneration within the hippocampus in this model. Three frames (1 × 1.2 mm per frame) encompassing a total area of 3.6 mm2 in each slide from each hippocampal CA1 region were analyzed at × 100 magnification. The mean number of degenerating neurons in the cortical and hippocampal CA1 regions from both the hemispheres in each group of animals was determined.
Statistical Analysis
ERK MAPK staining was semiquantitatively determined through the use of a 5-point scale (0, no stain; 5, maximal stain) by an investigator blinded to treatment. Pial artery diameter, CSF ERK MAPK, and degenerating neuron values were analyzed using analysis of variance for repeated measures. If the value was significant, the data were then analyzed by Fisher's protected least significant difference test. An α level of
Results
High-Dose tPA and RBC-tPA Cause Comparable Pial Artery Vasodilation After Cerebral H/I
Hypoxia/ischemia produces pial artery vasoconstriction (Armstead et al, 2008) and reduction in cerebral blood flow is thought to contribute to neuronal cell injury post the insult. The animals were treated for 30 mins before H/I (pretreatment) or 2 h post insult (post treatment) and the effect on the pial artery diameter induced by H/I was measured. Neither preinsult nor postinsult treatment with either tPA (0.1 mg/kg i.v.) or with the MAPK inhibitor U0126 (1 mg/kg i.v.) altered the effect of H/I on pial artery diameter (Figure 1). In contrast, RBC-tPA (0.1 mg/kg i.v.) or tPA (2 mg/kg i.v.) given preinsult or post insult caused a comparable increase in pial artery diameter in the setting of H/I (Figure 1). Red blood cell carriage enhanced the potency of tPA-induced vasodilation approximately 10-fold based on the dose administered.
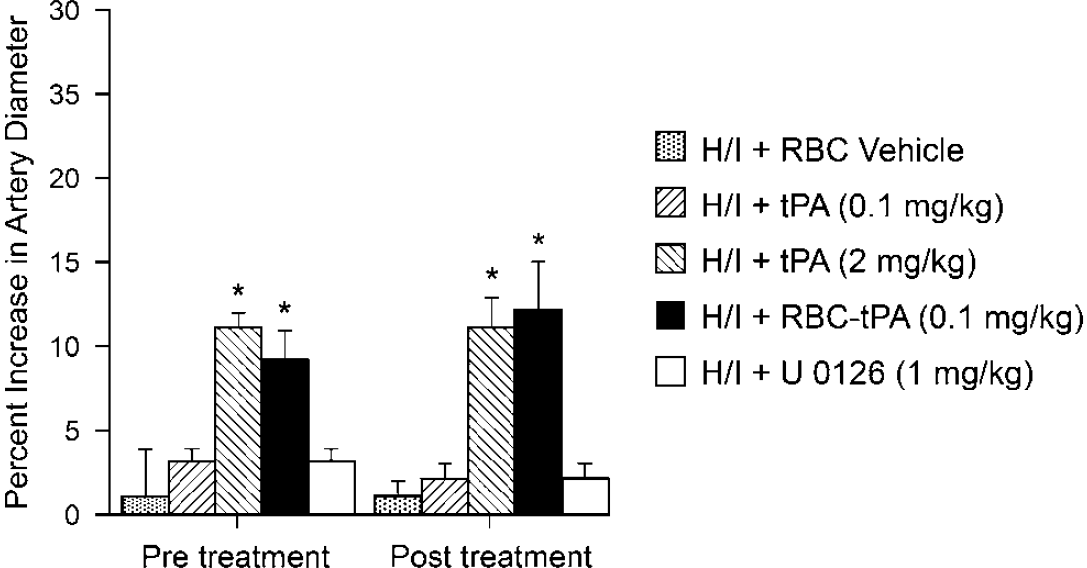
Influence of RBC vehicle, tPA (0.1 mg/kg i.v.), tPA (2 mg/kg i.v), RBC-tPA (0.1 mg/kg i.v.) and U0126 (1 mg/kg i.v.) on pial artery diameter after H/I,
RBC-tPA Prevents, Whereas tPA Aggravates, H/I-Induced Impairment of Hypercapnic and Hypotensive Pial Artery Dilation
Hypercapnia, hypotension, and isoproterenol elicited reproducible dilation of pial small arteries and arterioles (data not shown). Dilation of small pial arteries in response to hypercapnia was blunted after H/I, but reversed to vasoconstriction in pigs treated before (30 mins) or after (2 h) the insult with tPA (2 mg/kg i.v.) (Figure 2). Pretreatment or post treatment with either tPA (0.1 mg/kg i.v.) or RBC vehicle did not significantly affect the impaired responses to hypercapnia seen after H/I (Figure 2). In contrast, dilation was maintained at levels nearly equivalent to pre-H/I values in animals given RBC-tPA (0.1 mg/kg i.v.) preinsult or post insult. Similarly, the ERK MAPK inhibitor U0126 (1 mg/kg i.v.) preserved vasodilation in the setting of hypercapnia when given post insult (Figure 2).
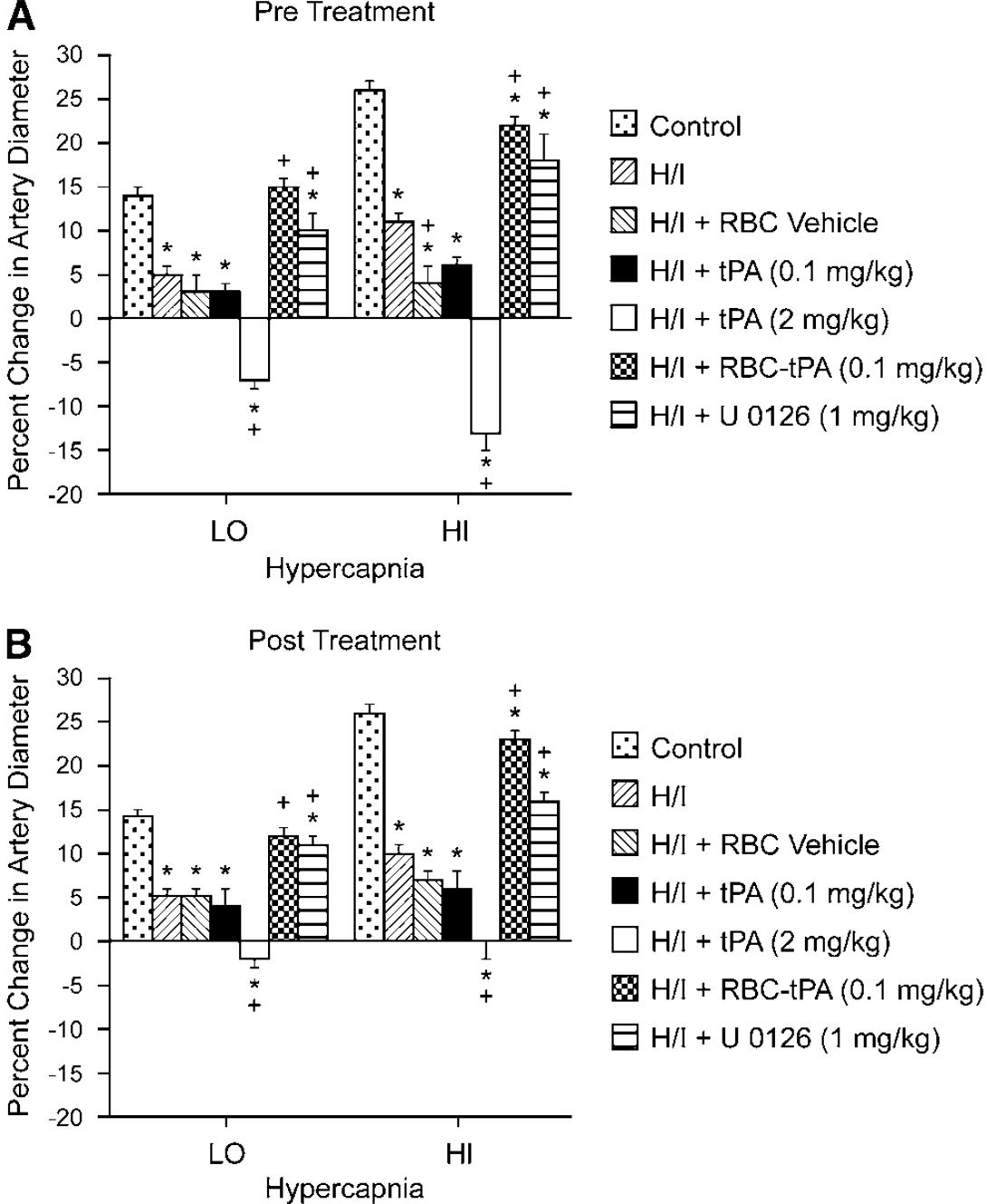
Soluble tPA administration aggravates H/I-induced impairment of hypercapnic pial artery dilation, whereas RBC-tPA prevents hypercapnic dilator impairment. Influence of hypercapnia (lo, hi; pCO2 of 50 to 55 and 70 to 75 mm Hg) on pial artery diameter before (control), 2.5 h after hypoxia/ischemia (H/I), after H/I treated with RBC vehicle, after H/I treated with tPA (0.1 mg/kg i.v), after H/I treated with tPA (2 mg/kg i.v), after H/I treated with RBC-tPA (0.1 mg/kg i.v), and after H/I treated with U0126 (1 mg/kg i.v),
Exogenous tPA (2 mg/kg i.v.) further impaired dilation of small pial arteries in response to the hypotension caused by H/I, whereas RBC-tPA (0.1 mg/kg i.v.), but not the RBC vehicle used to deliver tPA (0.1 mg/kg i.v.), and the ERK MAPK inhibitor both prevented (preinsult) or restored (post insult) vasodilation (Figure 3). Thus, RBC-tPA and tPA caused opposite effects on pial artery vasoreactivity impairment of hypercapnia and hypotension. It is of interest that pretreatment with tPA (2 mg/kg i.v.) impaired responses to these two stimuli to a greater extent than what was seen when tPA was given after the H/I insult (Figure 2 andFigure 3). These data indicate that prophylactic administration of tPA may produce a greater impairment of cerebral hemodynamics. Vasodilation of small pial arteries in response to isoproterenol was unaffected by H/I, tPA, RBC-tPA, and U0126 (Figure 4). Similar observations were made in pial arterioles (data not shown).
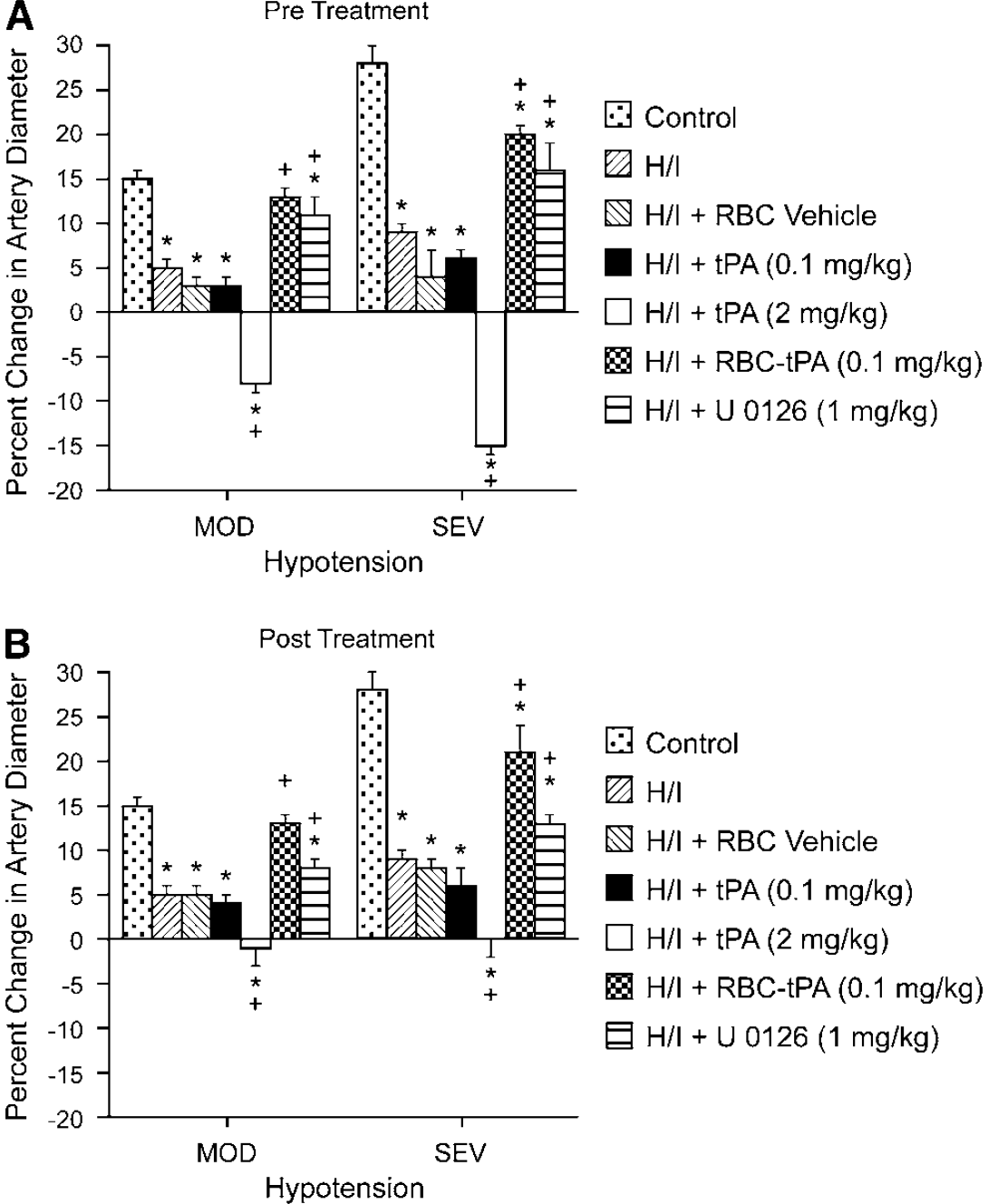
Soluble tPA administration aggravates H/I-induced impairment of hypotensive pial artery dilation, whereas RBC-tPA prevents hypotensive dilator impairment. Influence of hypotension (mod, sev; 25% and 45% reductions in mean arterial blood pressure for 10 mins) on pial artery diameter before (control), 2.5 h after hypoxia/ischemia (H/I), after H/I treated with RBC vehicle, after H/I treated with tPA (0.1 mg/kg i.v), after H/I treated with tPA (2 mg/kg i.v), after H/I treated with RBC-tPA (0.1 mg/kg i.v), and after H/I treated with U0126 (1 mg/kg i.v),
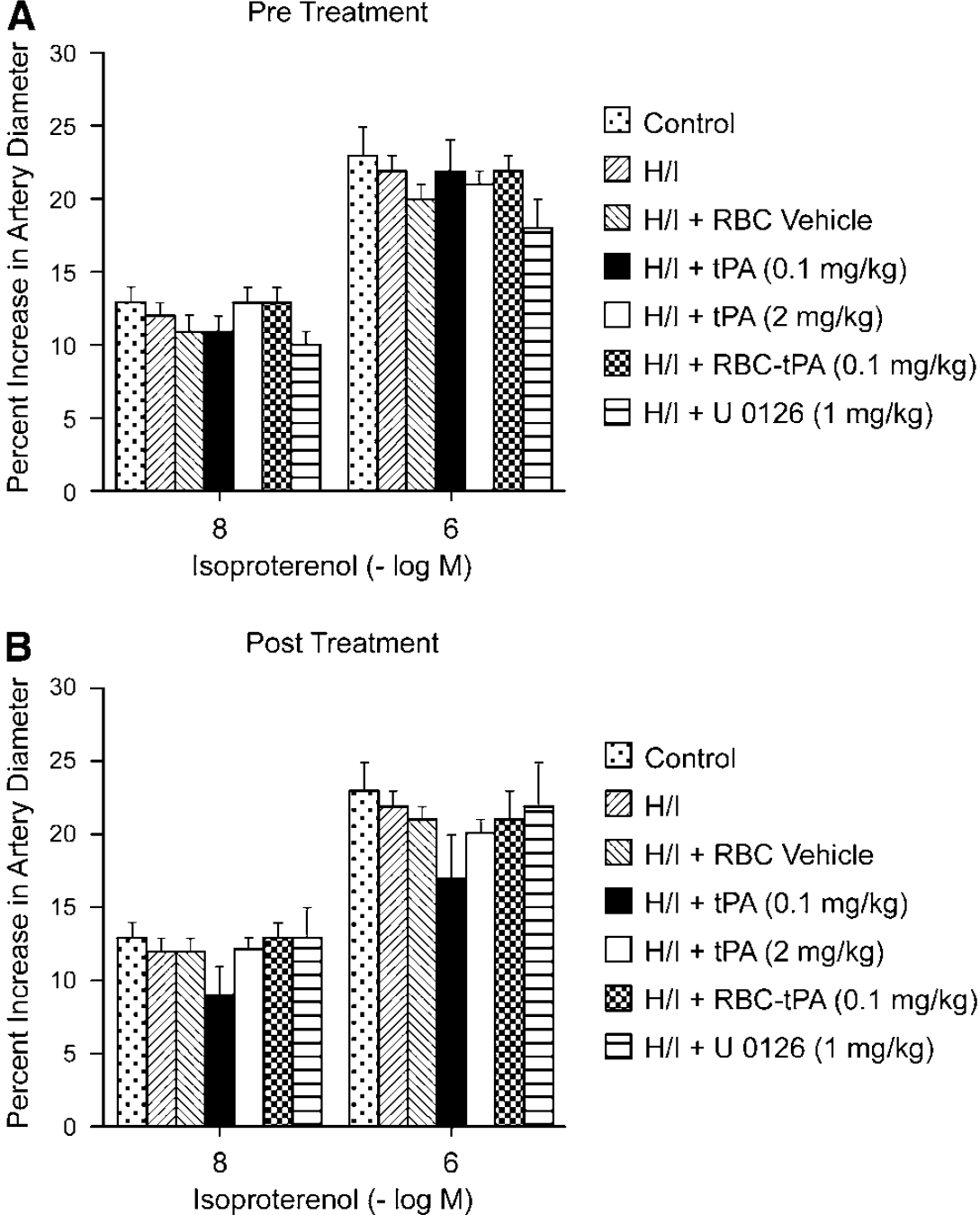
Soluble tPA and RBC-tPA have no effect on isoproterenol-induced pial artery dilation. Influence of isoproterenol (10−8, 10−6 mol/L) on pial artery diameter before (control), 2.5 h after hypoxia/ischemia (H/I), after H/I treated with RBC vehicle, after H/I treated with tPA (0.1 mg/kg i.v), after H/I treated with tPA (2 mg/kg i.v), after H/I treated with RBC-tPA (0.1 mg/kg i.v), and after H/I treated with U0126 (1 mg/kg i.v),
RBC-tPA Blunts, Whereas tPA Augments, H/I-Induced Phosphorylation of ERK MAPK
We next investigated the mechanism by which RBC-tPA preserved vasodilation beginning with its effect on phosphorylation of ERK MAPK after ischemia (Laher and Zhang, 2001). Cerebral H/I was induced in piglets. Two hours later the animals were given tPA (2 mg/kg i.v.), RBC-tPA (0.1 mg/kg i.v.), RBC vehicle, or U0126 (1 mg/kg i.v.). Two hours post treatment (4 h postcerebral H/I), phospho (activated) ERK MAPK was analyzed using an immunohistochemical approach (Figure 5). Abundant phospho ERK MAPK (4 to 5 on a 5-point scale) was observed in the hippocampus and the parietal cortex in animals given tPA after cerebral H/I (Figures 5 E and 5F). Tissue-type plasminogen activator increased the amount of phospho ERK MAPK compared with animals exposed to H/I that remained untreated (Figures 5C and 5D) (Armstead et al, 2008). The comparable section from a sham control animal was unremarkable (Figures 5A and 5B) (Armstead et al, 2008). In contrast, less phospho ERK MAPK was detected in the parietal cortex and the hippocampus of animals treated with RBC-tPA (Figures 5G and 5H) compared with animals exposed to H/I that were treated with tPA (Figures 5E and 5F). Detection of phospho ERK MAPK in animals treated with the RBC vehicle (lacking coupled tPA) (Figures 5K and 5L) was comparable to untreated animals post H/I (Figures 5C and 5D) (Armstead et al, 2008). U0126 blocked the upregulation of phospho ERK MAPK after H/I (Figures 5I and 5J). In contrast, no staining was seen with nonimmune IgG in the hypoxic/ischemic animal (Figure 5M insert).
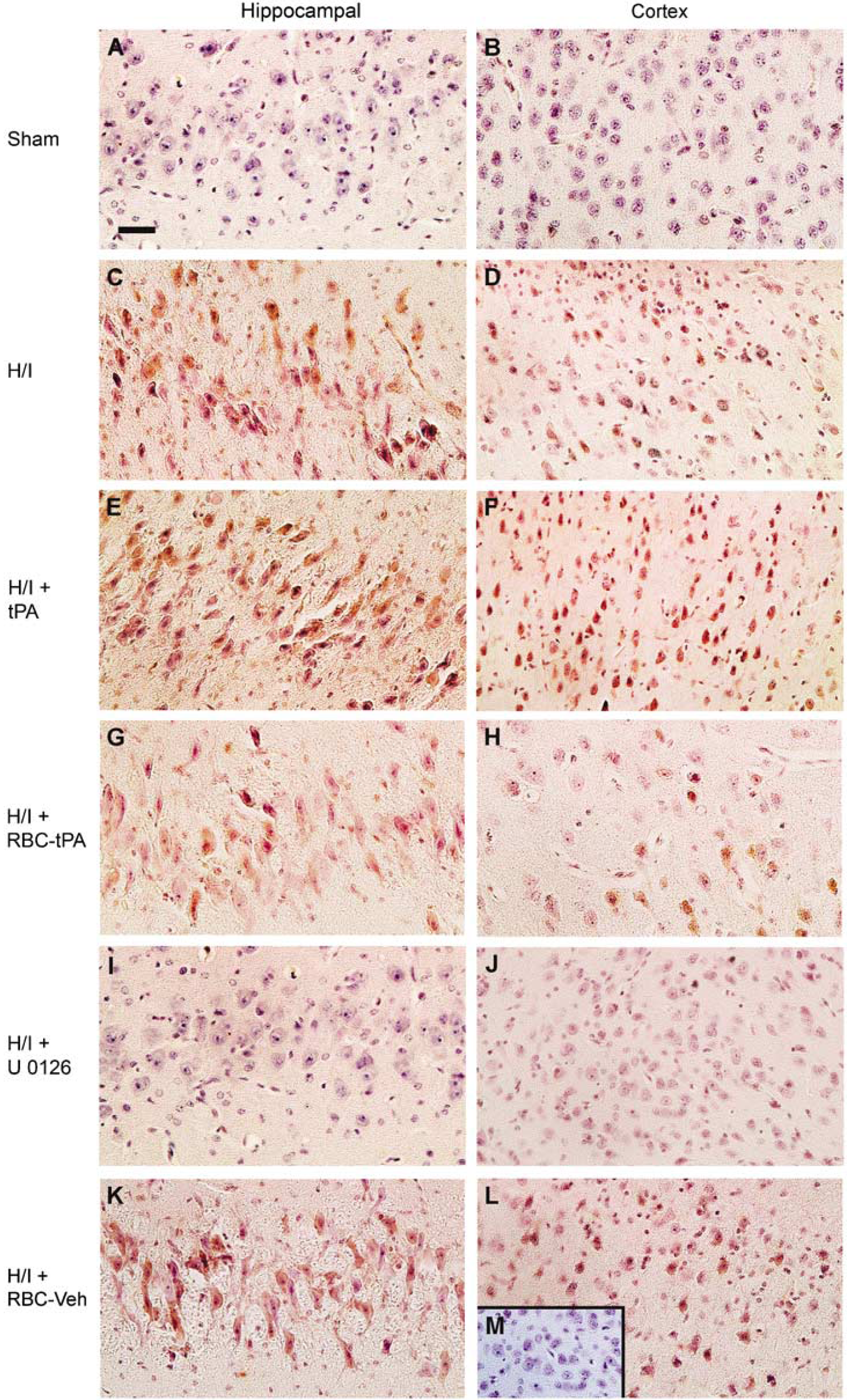
Tissue-type plasminogen activator aggravates H/I-induced ERK MAPK phosphorylation, whereas RBC-tPA blunts insult-associated increases in ERK MAPK phosphorylation. Immunohistochemical data for phospho (activated) ERK MAPK obtained from piglets 4 h after sham control (
To estimate ERK MAPK in the brain tissue, total and phosphorylated ERK MAPK in cortical periarachnoid CSF was measured by ELISA at baseline and after H/I. The activation (phosphorylation) state of the ERK MAPK isoform was determined by expressing the data as a percentage of control (total). Hypoxia/ischemia induced a marked phosphorylation (activation) of ERK MAPK within 1 h post injury (Figure 6). Exogenous tPA (2 mg/kg i.v.) administered 30 mins before, or 2 h after, H/I potentiated the phosphorylation of ERK MAPK (Figure 6). In contrast, administration of RBC-tPA (0.1 mg/kg i.v.) preinjury or postinjury blunted insult-induced phosphorylation of CSF ERK MAPK. It should be noted that RBC-tPA not only blocked the potentiation of CSF ERK MAPK release observed with tPA, but almost completely restored the values to those measured under sham control conditions (Figure 6). Administration of RBC vehicle or 0.1 mg/kg i.v. tPA preinsult or post insult failed to modify ERK MAPK phosphorylation (Figure 6). U0126 (1 mg/kg i.v.), a purported ERK MAPK antagonist, blocked ERK MAPK phosophorylation, as expected (Figure 6).
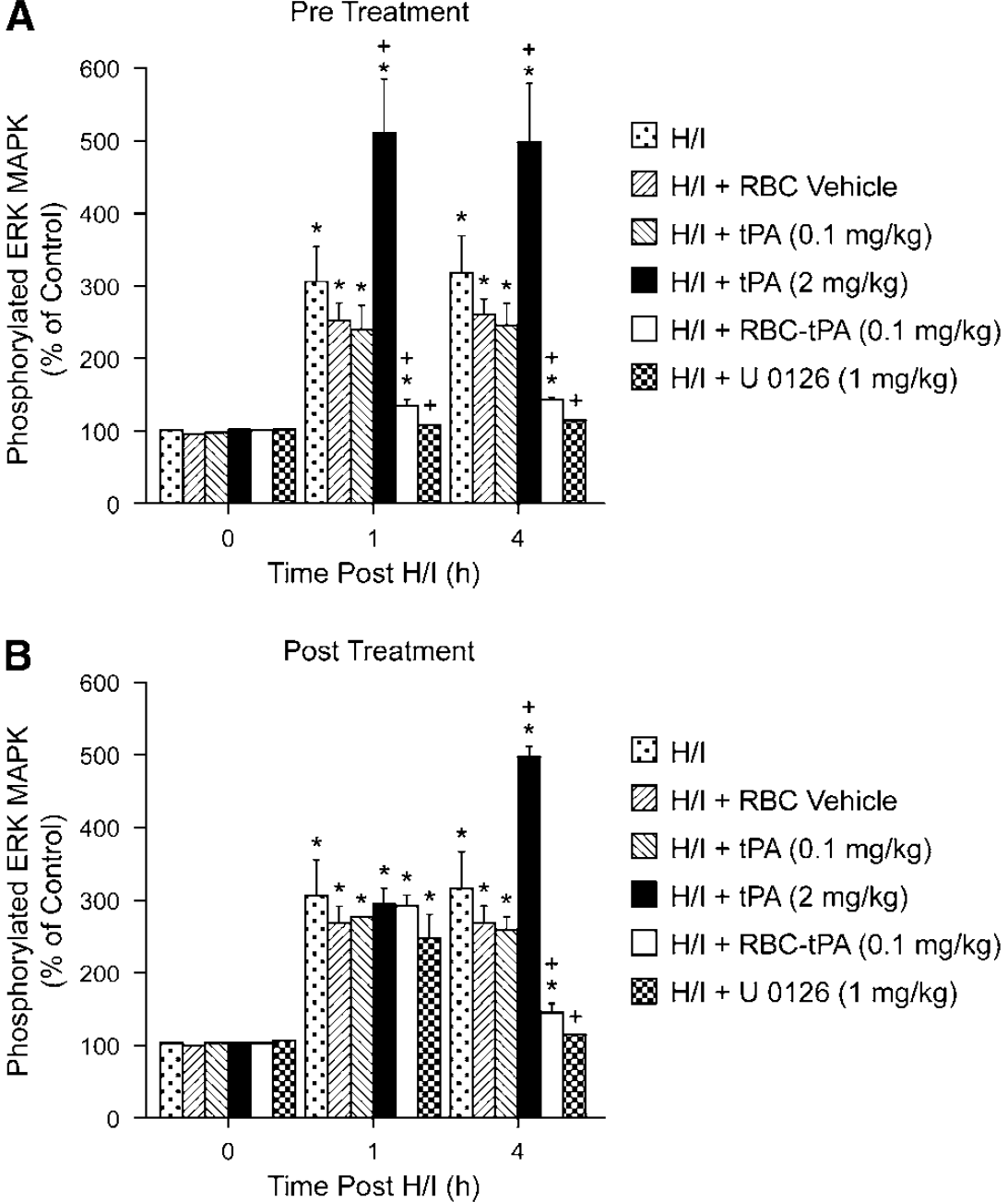
Phosphorylation of ERK MAPK in cortical periarachnoid CSF before H/I (0 min) and as a function of time (hour) after H/I in RBC vehicle, H/I and tPA (0.1 mkg/kg i.v.), in H/I + tPA (2 mg/kg i.v.), H/I + RBC coupled with tPA (0.1 mg/kg i.v), and H/I + U0126 (1 mg/kg i.v.) animals,
Post-Injury Treatment With RBC-tPA and U 0126 Attenuates Histopathologic Changes in the Parietal Cortex and the Hippocampus Associated With Hypoxia/Ischemia
Figure 7A shows HE staining for the hippocampal CA1 region and the parietal cortex under sham, H/I, H/I + tPA, H/I + RBC-tPA, H/I + RBC vehicle, and H/I + U0126 posttreated conditions 4h after cerebral H/I injury. Numerous necrotic neurons in the cortex and CA1 regions were seen in animals given tPA (2 mg/kg i.v.) after cerebral H/I (Figure 7A). In contrast, far fewer histopathologic abnormalities were seen in animals treated post H/I with RBC-tPA or U0126 (Figure 7A). Animals given the RBC vehicle evidenced large numbers of degenerating neurons, similar to those observed with H/I alone. The increase in the number of degenerating (pyknotic) neurons in the CA1 region after H/I was statistically significant (Figure 7B). Tissue-type plasminogen activator given after H/I caused greater neuronal necrosis than was seen in sham control animals (Figure 7B), but this value was not statistically greater than in untreated animals post H/I. Importantly, RBC-tPA and U0126 administration post insult caused a significant reduction in the number of degenerating neurons compared with animals treated with tPA (Figure 7B). Administration of tPA 30 mins before injury increased the severity of histologic damage compared with untreated H/I animals (Figure 7C). In contrast, administration of U0126 30 mins before injury ameliorated H/I-induced histologic damage (Figure 7C).
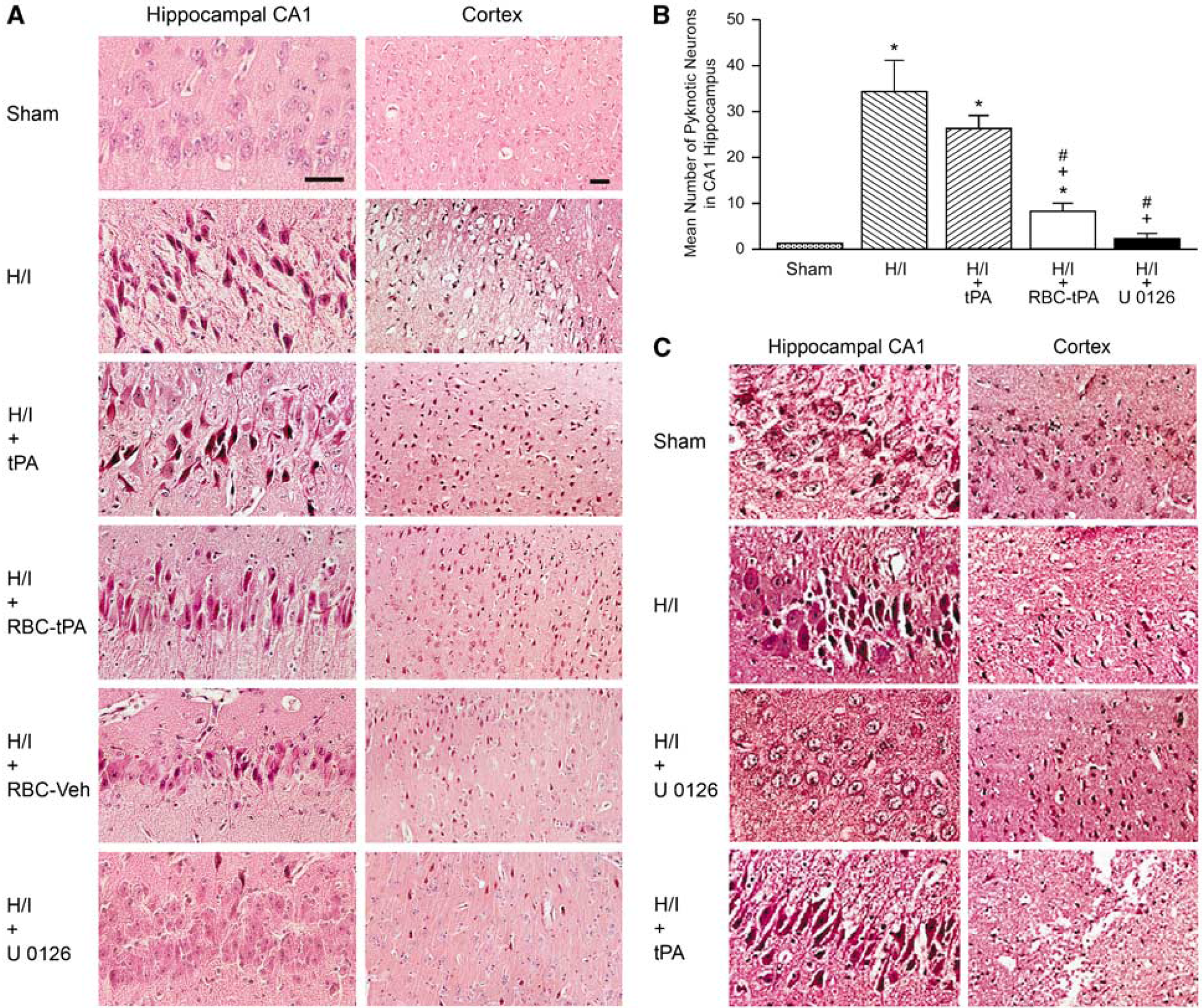
(
Blood Chemistry
Blood chemistry values were collected before and after all experiments. There were no statistically significant differences between sham control, H/I, and H/I and drug-treated animals. Hypoxia decreased pO2 to 35 ± 5 mmHg. Low levels of hypercapnia raised pCO2 to 58 ± 8 and high levels of hypercapnia raised pCO2 to 78 ± 9 mmHg. Carbon dioxide levels were kept constant during periods of hypoxia and oxygen levels were kept constant during periods of hypercapnia.
Discussion
The data in this paper show that coupling of tPA to carrier RBCs protects against cerebral injury caused when tPA is given in the setting of cerebral H/I as occurs in a stroke. Specifically, we found that the impairment of cerebrovascular responses to hypercapnia and hypotension associated with H/I was largely prevented by RBC-tPA. In earlier studies, we observed that pial artery dilation in response to these two stimuli was blunted post insult, reversed to pial artery vasoconstriction with the administration of tPA before the injury, and re-reversed to vasodilation with the plasminogen activator inhibitor-1 inhibitor, EEIIMD (Armstead et al, 2005b). As impaired responsiveness to cerebovasodilator stimuli is thought to contribute to an adverse outcome, RBC-tPA may offer a novel approach toward the treatment of CNS injury associated with H/I.
These findings have been extended in this study by the observation that tPA and RBC-tPA have diametrically opposite effects on the responsiveness of pial arteries to vasodilator stimuli post insult. Administration of tPA (2 mg/kg i.v.) 2 h post injury exacerbates impairment of cerebrovascular responses to hypercapnia and hypotension. In contrast, administration of RBC-tPA (0.1 mg/kg i.v.) given at the same time in the same setting prevents impairment of dilation to hypercapnia and hypotension post insult. These data indicate that coupling of tPA to RBC enhances its provasodilation effect in the piglet CNS exposed to H/I, likely because of the prolongation of the half-life in the circulation and retention in the intravascular compartment. As tPA and RBC-tPA elicited comparable pial artery vasodilation, RBC carriage enhanced the potency of tPA-induced vasodilation approximately 10-fold based on the dose administered. The protective effect on stimulus-induced vasodilation provided by RBC-tPA appears to be selective for several reasons. First, neither did coupling of the vehicle cause vasodilation nor did it prevent impairment in dilator responses post insult. Second, cerebral H/I does not exert a nonspecific global impairment in the vasculature as responses to isoproterenol were unchanged post insult, as reported earlier (Armstead et al, 2005b, 2008). Third, RBC-tPA did not alter the dilation in animals administered isoproterenol, excluding a nonspecific enhancement of cerebrovasodilator responsiveness. The observation that RBC-tPA preserves cerebrovasodilation whether given pre-H/I or post H/I suggests its utility both as a treatment for cerebral ischemia and as a means to prevent delayed injury in settings in which the risk of recurrent ischemic occlusion is high.
Three additional conclusions can be drawn from our findings. First, the clinical relevance of preserving cerebral vasodilation is supported by the finding that administration of RBC-tPA at a 10-fold lower dose pre-H/I or post H/I reduced neuronal degeneration in the CA1 hippocampus whereas tPA had no protective effect, although our studies do not formally prove that preservation of vasodilation promotes blood flow and thereby prevents neuronal injury. Second, the neuronal cell loss between animals treated with tPA, RBC-tPA, or not treated at all, indicates that spatial containment of tPA within the vasculature limits extravasation into the brain parenchyma to promote neuronal cell injury. Third, the data also help affirm our earlier findings that tPA regulates the cerebral vascular tone by acting within the vasculature (Armstead et al, 2005b, 2008).
Blockade of ERK MAPK upregulation preserved vasodilation in animals that were given RBC-tPA. This extends earlier findings that indicate that upregulation of ERK MAPK impairs responses to stimulus-induced cerebrovasodilation (Armstead et al, 2008). The plasminogen activator induced vascular activity and signal transduction is thought to be mediated at least, in part, through LRP (Armstead et al, 2008; Bu et al, 1992). Hypoxia/ischemia-associated upregulation of ERK MAPK is potentiated by exogenous urokinase plasminogen activator and inhibited by the LRP antagonist RAP, an anti-LRP-1 antibody, and by U0126, a purported ERK MAPK selective antagonist (Armstead et al, 2008). U0126 functions as an ERK MAPK antagonist in this experimental model based on Western analysis and ELISA (Armstead et al, 2008). The immunohistochemical and quantitative ELISA data in this study confirm and extend earlier observations by showing that the injury caused by administering tPA before or after cerebral H/I is associated with the upregulation of ERK MAPK in the cerebral cortex and the hippocampus. In contrast, RBC-tPA decreased upregulation of ERK MAPK and attenuated injury. The specificity of this relationship is supported by the findings that U0126 blocked the increased expression of ERK MAPK post H/I, whereas the vehicle for the RBC conjugate had no effect. An unexpected finding was that the RBC-tPA actually reduced the upregulation of ERK MAPK to the level seen under sham control conditions. By inference, spatial constraint of tPA on the vasculature might limit the ability of tPA to interact with LRP and/or cells in the brain parenchyma in which ERK MAPK is upregulated. Alternatively, the lower dose of tPA used when coupled with RBC may also have contributed to the result observed. Whether the MAPK isoform expression profile is also altered is under investigation. We hypothesize that the capacity of the RBC linkage to attenuate upregulation of ERK MAPK expression by tPA provides a mechanistic link between cerebral hemodynamic responsiveness and neuronal cell integrity in the setting of cerebral H/I. However, additional study will be needed to establish a cause—effect relationship.
The dose of free tPA was selected based on the one used clincally (1 mg/kg). Review of the literature shows the range of therapeutic doses of tPA in humans and other included species to be within 1 to 10 mg/kg. Relatively higher doses in rats and mice are necessitated by the less efficient fibrinolysis in the blood of these species because of enzyme/substrate incongruency between human tPA and rodent plasminogen, as well as the reported higher activity of plasma inhibitors, including plasminogen activator inhibitor-1 (Ganguly et al, 2005). Our studies of
Many studies of cerebral ischemia have been performed in rodent models. Piglets offer a unique advantage in elucidating pathways involved in CNS ischemic injury by virtue of having a gyrencepahalic brain that contains substantial white matter similar to humans, and which is more sensitive to ischemic damage than gray matter (Shaver et al, 1996). On the basis of interspecies extrapolation of brain growth curves (Dobbing, 1974), the age of the newborn pig used in these studies roughly approximated the newborn infant time period in the human. Although the incidence of cerebral ischemic events in the pediatric population is relatively low compared with the adult (Janjua et al, 2007), the magnitude of the condition is amplified by the potential long-term loss of quality-of-life years for children with CNS ischemic disorders.
Indeed, the 2001 workshop report of the National Institute of Neurological Disorders and Stroke noted a deficiency in research in pediatric stroke related to the paucity of animal models and basic research investigation into ischemic disorders of the CNS in the pediatric population (Lynch et al, 2002). The use of a piglet model importantly allows for the study of physiologic variables and is more clinically relevant than the rodent models. The identification of a molecular target (ERK MAPK) for modifying neuropathology injury is in itself an important observation and potentially clinically relevant as there are, to date, no clinically proven neuroprotective interventions other than hypothermia for neonatal hypoxic/ischemic brain injury. The findings in this study regarding tPA are equally important as more attention is focused on treating stroke in infants and children. The important finding is that tPA alone may exacerbate neuronal injury in the setting of H/I. This observation was made in adult models of focal arterial-occlusive stroke years ago, but its significance was diminished, indeed outweighed, by the overwhelming clinical trial evidence of benefit because of fibrnolysis. However, the use of tPA for adult acute stroke is highly constrained so as to minimize the risk of hemorrhage. To date there have been no safety or efficacy trials for tPA in the treatment of infants and children with acute stroke. Many individual children, however, are receiving tPA based on the assumption that studies in adults are generalizable to children. The data from this study provide additional evidence that both safety and efficacy of tPA—whether it be traditional i.v. or intrarterial tPA, or RBC-tPA—must be evaluated systematically in children before being widely adopted in clinical care.
As neonates are at risk for intraventricular hemorrhage, it could be seen as counterintuitive to administer a plasminogen activator. However, the fact that endogenous plasminogen activator release occurs after cerebral H/I (Armstead et al, 2008) makes our nonthrombotic model relevant to the investigation of the regulation of cerebrovasodilator mechanisms and histopathology.
Using a model dominated by thrombosis could well be criticized as likely to yield ambiguous results, as the salutary effects of fibrinolysis will be variably offset by the deleterious effects of tPA on vascular tone depending on the specific experimental setting. We seek to delineate the mechanism by which fibrinolytic therapy causes CNS damage and to help re-engineer these molecules through use of novel delivery systems or use selective inhibitors of the signal transduction pathway(s) they initiate to increase their benefit/risk ratio. The nonthrombotic model of cerebral H/I is well suited for this purpose. Nonetheless, clinically, cerebral H/I is often associated with neonatal stroke (Ferriero, 2004). Therefore, investigation of the potential for tPA to have deleterious effects in the setting of cerebral H/I and their amelioration with RBC-tPA, in a piglet model might have considerable translational relevance. Although the present studies might have implications for the pediatric population, our results may also have wider applicability given the observed beneficial action of RBC-tPA in adult animal models of stroke and TBI (Danielyan et al, 2008; Stein
In summary, our results indicate a protective effect of coupling of tPA to RBC in the setting of cerebral ischemia. The observation that RBC-tPA preserves cerebrovasodilation whether given pre-H/I or post H/I suggests its utility both as a treatment for cerebral ischemia and a means to prevent further injury in settings in which the risk of recurrent ischemic occlusion is high. Our studies suggest that RBC-tPA may increase the benefit ratio of thrombolytic interventions and extend the benefits to additional patients in the pediatric age group with stroke and ischemic disorders involving the CNS.
Conflict of interest
The authors declare no conflict of interest.