Nitric oxide (NO) contributes to hypoxia-induced pial artery dilation, at least in part, through the formation of cGMP and the subsequent release of methionine enkephalin and leucine enkephalin in the newborn pig. In separate studies, these opioids also were observed to elicit NO-dependent pial artery dilation, whereas light/dye endothelial injury reduced hypoxic pial dilation. The current study was designed to investigate the role of the endothelial isoform of NO synthase in hypoxic pial dilation, associated opioid release, and opioid dilation in piglets equipped with a closed cranial window. N-iminoethyl-l-ornithine (l-NIO) (10−6 mol/L), an antagonist that may have greater endothelial NO synthase inhibitory selectivity, had no effect on dilation elicited by hypoxia (Po2 ≈ 35 mm Hg) (24 ± 2 versus 24 ± 2% in the absence and presence of l-NIO, respectively, n = 8). Hypoxic dilation was accompanied by increased CSF cGMP, which also was unchanged in the presence of l-NIO (394 ± 19 and 776 ± 63 versus 323 ± 13 and 739 ± 25 fmol/mL for control and hypoxia in the absence and presence of l-NIO, respectively, n = 6). Additionally, hypoxic pial dilation was associated with increased CSF methionine enkephalin, which also was unchanged in the presence of l-NIO (992 ± 73 and 2469 ± 197 versus 984 ± 18 and 2275 ± 185 pg/mL, respectively, n = 6). In contrast, methionine enkephalin–induced dilation was blocked by l-NIO (6 ± 1, 10 ± 1, and 16 ± 1 versus 1 ± 1, 1 ± 1, and 2 ± 1% for 10−10, 10−8, 10−6 mol/L methionine enkephalin, respectively, before and after l-NIO, n = 8). Substance P–induced pial dilation was blunted by l-NIO, whereas responses to sodium nitroprusside and N-methyl-d-aspartate were unchanged. These data indicate that endothelial NO synthase contributes to opioid-induced pial artery dilation but not hypoxia-induced dilation. Additionally, these data suggest that neuronally derived NO contributes to hypoxic pial dilation.
Numerous mechanisms for hypoxia-induced cerebral vasodilation have been proposed, including adenosine, prostaglandins, and vasopressin (Coyle et al., 1993; Emerson and Raymond, 1981; Eisenach et al., 1992). Previous studies have examined the role of nitric oxide (NO) in cerebrovasodilation elicited by hypoxia. For example, Iwamoto and others (1992) observed that N-nitro-l-arginine (l-NNA) attenuated the increase in cerebral blood flow during hypoxia in the adult sheep. Conversely, Kozniewska and others (1992) reported that systemic administration of the NO synthase inhibitor, N-mono-methyl-l-arginine does not alter the increase in cerebral blood flow elicited by hypoxia in the adult rat. In the newborn pig, NO has been observed to both contribute to hypoxic pial dilation and not contribute to hypoxic hyperemia (Armstead, 1997b, 1995b; Ichord et al., 1994). These data indicate that there is a case, although controversial, for NO involvement. Discrepancies between these studies could result from differences in doses or timing of antagonists administered, the species or the age of the animal investigated, or effect of antagonist on the oxyhemoglobin dissociation curve (Pelligrino et al., 1995).
Other studies have examined the role of opioids in hypoxic cerebrovasodilation. For example, it was observed that hypoxia increases plasma methionine enkephalin in fetal sheep (Martinez et al., 1994) and plasma β endorphin in human newborns at delivery (Sankaran et al., 1983) and in infants with hypoxic–ischemic encephalopathy with ongoing hypoxia (Martinez et al., 1990). In the piglet, hypoxia increases cortical periarachnoid CSF methionine enkephalin and leucine enkephalin concentration, whereas antagonists of these opioids diminish hypoxic pial artery dilation, indicating their involvement in the vascular response to the stimulus (Armstead, 1995a,
b
).
In addition to acting separately, NO and opioids also interact in the modulation of hypoxic pial dilation in the piglet. For example, sodium nitroprusside (SNP) increases CSF methionine enkephalin and leucine enkephalin through the formation of cGMP (Wilderman and Armstead, 1996). Additionally, hypoxic elevation of the CSF concentration for both opioids was attenuated by l-NNA, suggesting that NO contributes to hypoxic dilation, at least partially, by the formation of cGMP and the subsequent release of opioids. In separate studies, methionine enkephalin and leucine enkephalin also were observed to elicit dilation in a NO-dependent manner (Devine and Armstead, 1995). Since NO therefore contributes to both the release of opioids as well as their subsequent vascular response, these data may create a feed-forward loop. If the pools of NO used for vasodilation and opioid release were compartmentalized, however, such a situation may not occur.
Vascular tone is regulated by NO production through two constituitively expressed isoforms of NO synthase: neuronal and endothelial (Fostermann et al., 1994). Basal-stimulated and pharmacologically stimulated endothelial NO synthase activity contributes to the regulation of vascular tone and accounts for many actions of endothelium derived relaxing factor (Moncada et al., 1991). Alternatively, evidence for the contribution of neuronal NO to the control of cerebral hemodynamics also has been documented (Kelly et al., 1995; Wang et al., 1995). Since the light/dye endothelial injury technique has been observed to reduce pial artery dilation to hypoxia in the piglet (Leffler et al., 1997), the endothelial isoform of NO may contribute to that response. However, the available pharmacologic tools have not allowed the dissection of the relative contributions of the neuronal and endothelial NO isoform to a particular response with any confidence. N-iminoethyl-l-ornithine (l-NIO) has been suggested to have greater endothelial NO synthase inhibitory selectivity (Rees et al., 1990), and therefore may be a useful probe for characterizing the contribution of endothelial NO synthase to the control of the cerebral circulation.
The current study was, therefore, designed to investigate the role of the endothelial isoform of NO synthase in hypoxic pial artery dilation, associated opioid release, and opioid-induced vasodilation.
METHODS
All experiments have been approved by the Institutional Animal Care and Use Committee. Pigs (1 to 5 days old) of either sex were used in these experiments. They were first anesthetized with ketamine hydrochloride-acepromazine (33 mg/kg intramuscularly). Anesthesia was maintained with α-chloralose (30 to 50 mg/kg initially, supplemented with 5 mg/kg intravenously). A catheter was inserted into the femoral artery to record blood pressure and to sample for blood gases and pH. Another catheter was placed in a femoral vein for injection of drugs. The trachea was cannulated, and the animals were ventilated with room air. The body temperature was maintained at 37 to 38°C with a heating pad.
For insertion of the cranial window, the scalp was removed and an opening was made in the skull over the parietal cortex. The dura was cut and retracted over the cut bone edge. The cranial window was placed in the hole and cemented in place with dental acrylic. The space under the window was filled with artificial CSF of the following composition (mg/mL): 220 KCl, 132 MgCl2, 221 CaCl2, 7720 NaCl, 402 urea, 665 dextrose, and 2066 NaHCO3 (with pH 7.30 to 7.36, Pco2 42 to 49 mm Hg, and Po2 40 to 50 mm Hg).
Pial arterioles were observed with a dissecting microscope, a television camera mounted on the microscope, and a video monitor. Vascular diameter was measured with a video microscaler.
Protocol
Pial artery diameter (small artery 120 to 160 μm; arteriole 50 to 70 μm) was determined every minute for a 10-minute exposure period after injection under the window of artificial CSF containing no drug and CSF containing a drug. Diameters also were measured 10 to 15 minutes after the highest concentration of a drug was flushed off the cerebral cortical surface with CSF containing no drug. Typically, 1 to 2 mL of CSF were flushed through the window over a 30-second period. Needles incorporated into the side of the window allowed for the injection of CSF under the window and the runoff of excess CSF. The peak responses were measured, and a CSF sample for vasoactive metabolite analysis was collected at the end of the 10-minute exposure period. The cerebral cortical periarachnoid CSF (300 μL) was collected slowly by infusing artificial CSF into one side of the window and allowing the CSF under the window to drip freely into a collection tube on the opposite side.
Responses were observed after the administration of methionine enkephalin and leucine enkephalin (10−10, 10−8, 10−6 mol/L; Sigma Chemical Co., St. Louis, MO) and in the absence and presence of l-NIO (10−6 mol/L; Tocris Cookson, St. Louis, MO), a purported endothelial NO synthase inhibitor. Responses also were observed after administering SNP, N-methyl-d-aspartate (NMDA), and substance P (10−8, 10−6 mol/L, Sigma) in the absence and presence of the antagonist, l-NIO. Animals received a maximum of three agonists, each at two or three concentrations, administered in ascending concentration fashion before and after one antagonist. The antagonist was placed on the brain for 10 minutes before its coadministration with an agonist. To determine if dilator responses to SNP were dependent on the release of opioids, dilation by SNP was observed before and after the administration of the opioid antagonist, naloxone (1 mg/kg intravenously, Sigma). Time control experiments were designed so that responses were obtained initially (defined as first in the tables) and then again 60 minutes later (defined as second).
Hypoxia was produced by decreasing the inspired oxygen sufficiently to reduce and maintain arterial Po2 at 35 ± 3 mm Hg (for moderate hypoxia) and 25 ± 3 mm Hg (for severe hypoxia) while maintaining constant arterial Pco2 in the normocapnic range (33 ± 3 mm Hg). Changes in pial artery diameter were measured every minute during the 10-minute hypoxic exposure period. A sample of blood confirming the hypoxia was taken 3 to 4 minutes after the hypoxia began. Once the blood chemistry data confirmed that the desired level of hypoxia had been achieved, peak dilator responses were recorded. Responses to hypoxia were obtained before and after topical applications of l-NIO. For hypoxia responses in the presence of l-NIO, the inhibitor was topically applied 10 minutes before induction of hypoxia, and the subsequent effects of the inhibitor on hypoxia-induced pial artery dilation were observed for the succeeding 10 minutes.
Appropriate aliquots of the vehicles for all agents (0.9% saline) were added to CSF infused under the window. These CSF vehicles had no effect on artery diameter. All working drug solutions were made fresh on the day of use.
Opioid analysis
The CSF samples collected were acidified with 1 N acetic acid to prevent protein degradation and stored at -20°C. Radioimmunoassay kits for methionine enkephalin and leucine enkephalin are commercially available (IncStar, Stillwater, MN; Peninsula Laboratory, Belmont, CA, U.S.A.). The radioimmunoassay used simultaneous addition of the sample, antiopioid, and the 125I derivative of the opioid. After an overnight incubation at 4°C, the free opioid was first separated from the opioid bound to the antibody, by the addition of saturated ammonium sulfate in the presence of rabbit carrier γ-globulin. After centrifugation at 760 g for 10 minutes, the supernate was decanted and the pellet was counted using a gamma scintillation counter. All samples and standards were assayed in duplicate. Data were calculated at %B/Bo versus concentration, where B/Bo is calculated as follows: (average counts per minute of sample – the average counts per minute of nonspecific binding tube)/(average counts per minute of total binding tube – the average counts per minute of nonspecific binding tube) × 100.
Cyclic nucleotide analysis
Cerebrospinal fluid samples collected after a 10-minute exposure to a drug were analyzed for cGMP concentration using scintillation proximity assay methods. Commercially available kits for cGMP (Amersham, Arlington Heights, IL, U.S.A.) were used. Briefly, the assay determines cyclic nucleotide concentration for binding to antiserum that has a high specificity for the cyclic nucleotide. The antibody-bound cyclic nucleotide then is reacted with an anti-rabbit second antibody bound to fluromicrospheres. Labeled cyclic nucleotide bound to the primary rabbit antibody then can be measured by determining the amount of light emitted by the fluoromicrospheres. All unknowns are assayed at two dilutions. The concentration of the unlabeled cyclic nucleotide is calculated from the standard curve using linear regression analysis.
Statistical analysis
All measures were analyzed using analysis of variance for repeated measures. If the values were significant, the Fisher test was performed. An a level of P<0.05 was considered significant in all statistical tests. Then n values reflect data for one vessel in each animal. Values are represented as means ± standard deviation of absolute values or as percentages of changes from control values. Data presented as percentage change were compared by nonparametric means using the Wilcoxon signed rank test.
RESULTS
Role of endothelial NO synthase in hypoxia-induced pial artery dilation and opioid release
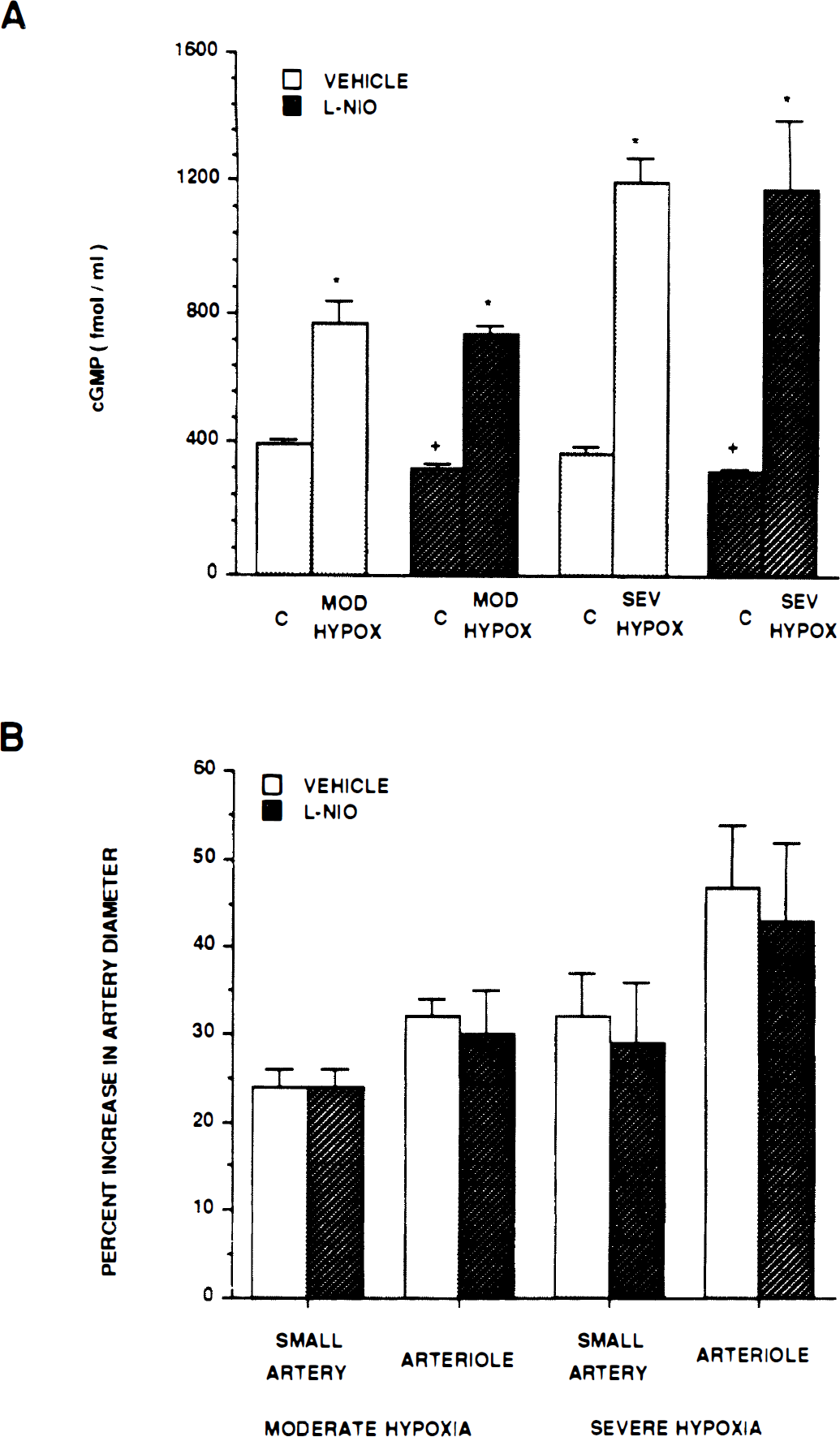
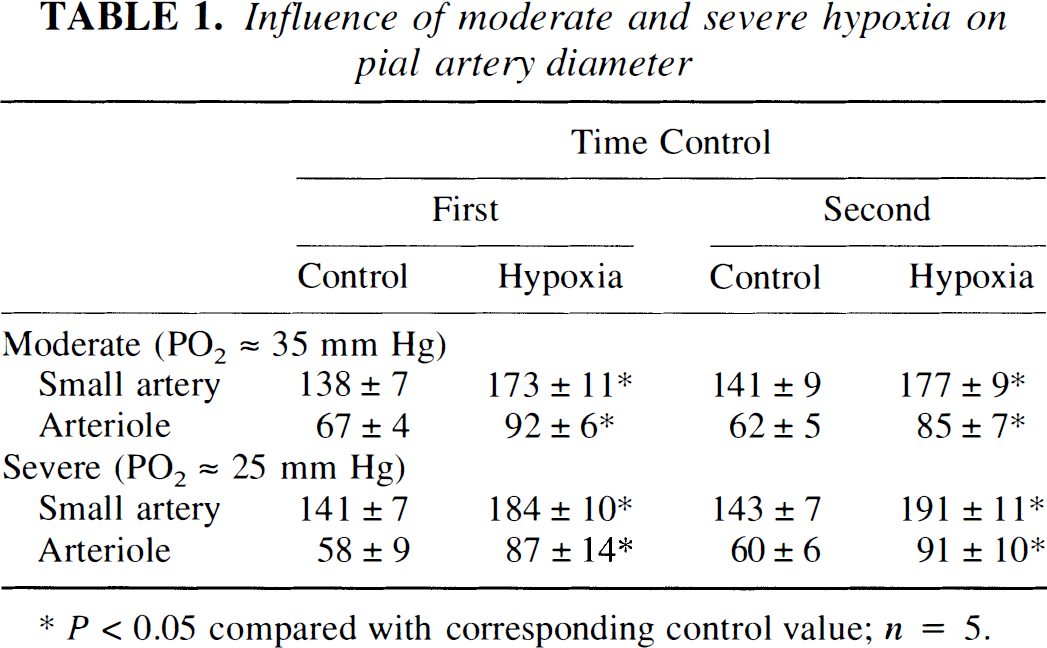
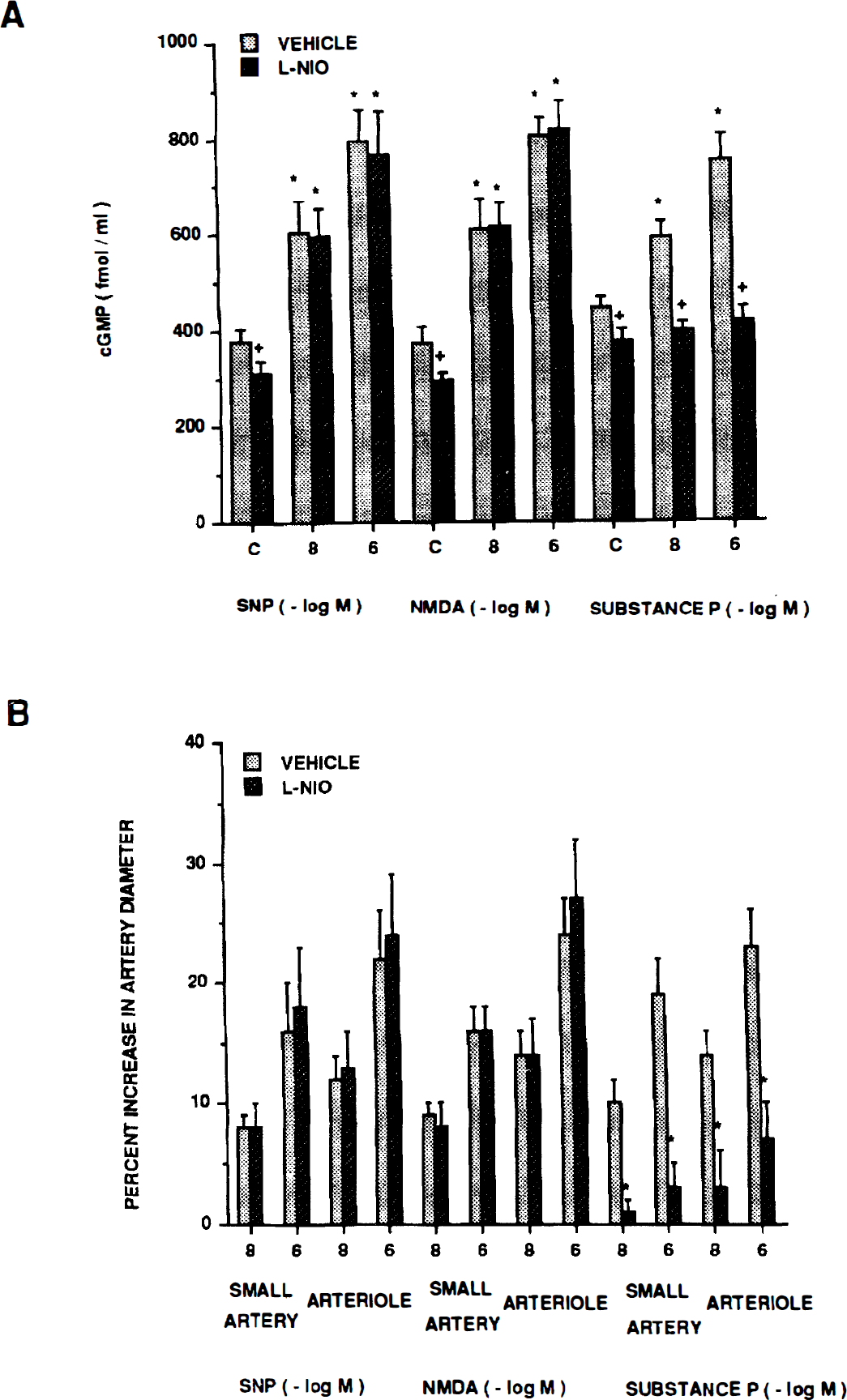
Dilation in response to moderate and severe hypoxia elicited reproducible pial small artery (120 to 160 μm) and arteriole (50 to 70 μm) vasodilation (Table 1). Dilation caused by hypoxia was unchanged by l-NIO (10−6 mol/L) (Fig. 1). Increases in cortical periarachnoid CSF cGMP, methionine enkephalin, and leucine enkephalin associated with hypoxia also were unchanged in the presence of l-NIO (Fig. 1 and Fig. 2).
(A) Influence of moderate hypoxia (Po2 ≈ 35 mm Hg) and severe hypoxia (Po2 ≈ 25 mm Hg) on cortical periarachnoid CSF cGMP concentration (fmol/mL) in the absence (vehicle) and presence of N-iminoethyl-l-omithine (l-NIO) (10−6 mol/L). Value are means ± SD, n = 6. *P<0.05 compared with corresponding control value (c). +P<0.05 compared with corresponding vehicle value. (B) Influence of moderate hypoxia and severe hypoxia on small pial arteries and arterioles in absence (vehicle) and presence of l-NIO (10−6 mol/L). Values are mean ± SD; n = 8 for moderate hypoxia, n = 12 for severe hypoxia. Mod Hypox, moderate hypoxia; Sev Hypox, severe hypoxia.
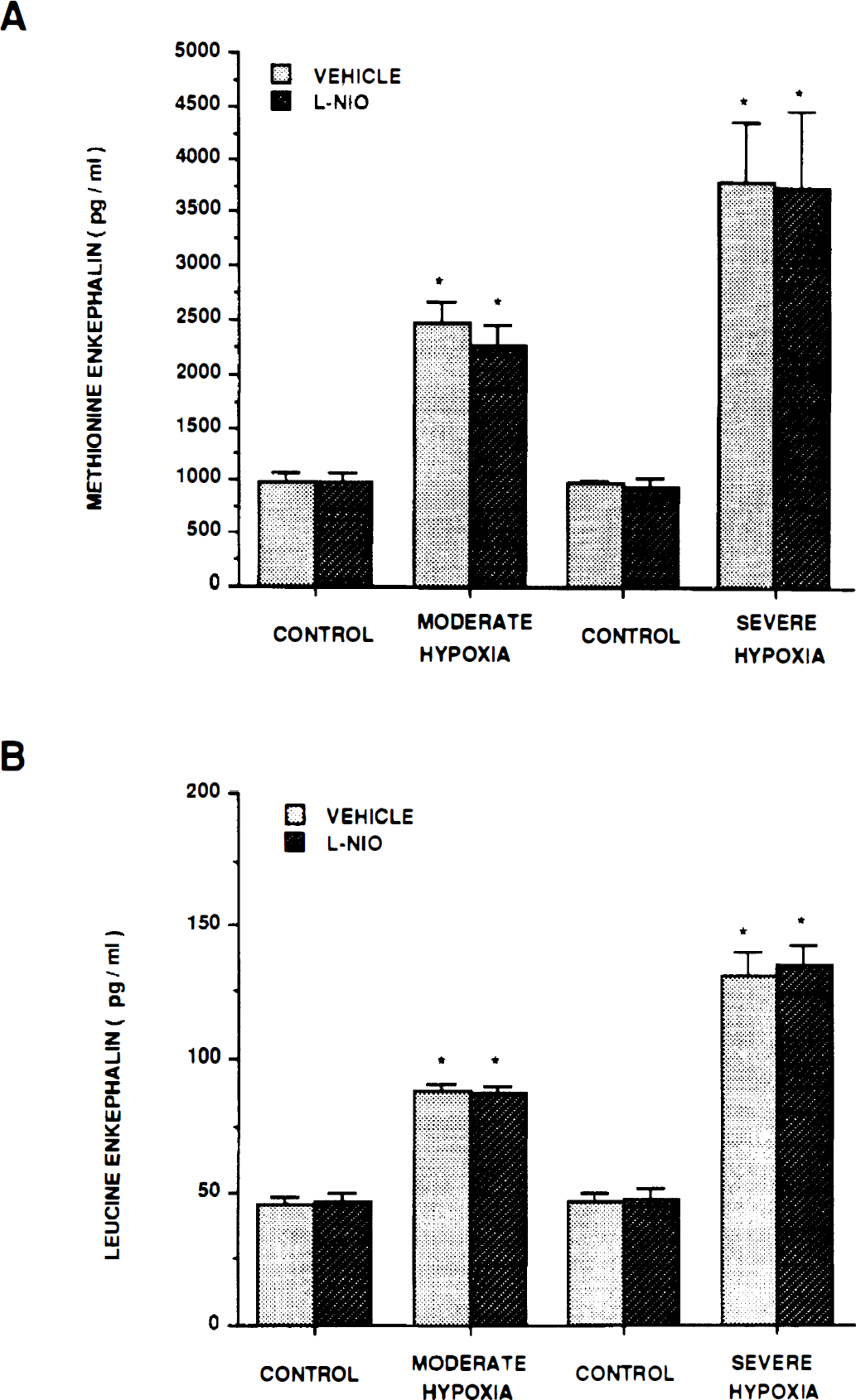
Influence of moderate hypoxia Po2 ≈ 35 mm Hg) and severe hypoxia (Po2 ≈ 25 mm Hg) on CSF opioid concentration (pg/mL) in the absence (vehicle) and presence of N-iminoethyl-l-omithine (l-NIO) (10−6 mol/L). Methionine enkephalin (A); leucine enkephalin (B). Values are mean ± SD, n = 6. *P<0.05 compared with corresponding control value.
Influence of moderate and severe hypoxia on pial artery diameter
P<0.05 compared with corresponding control value; n = 5.
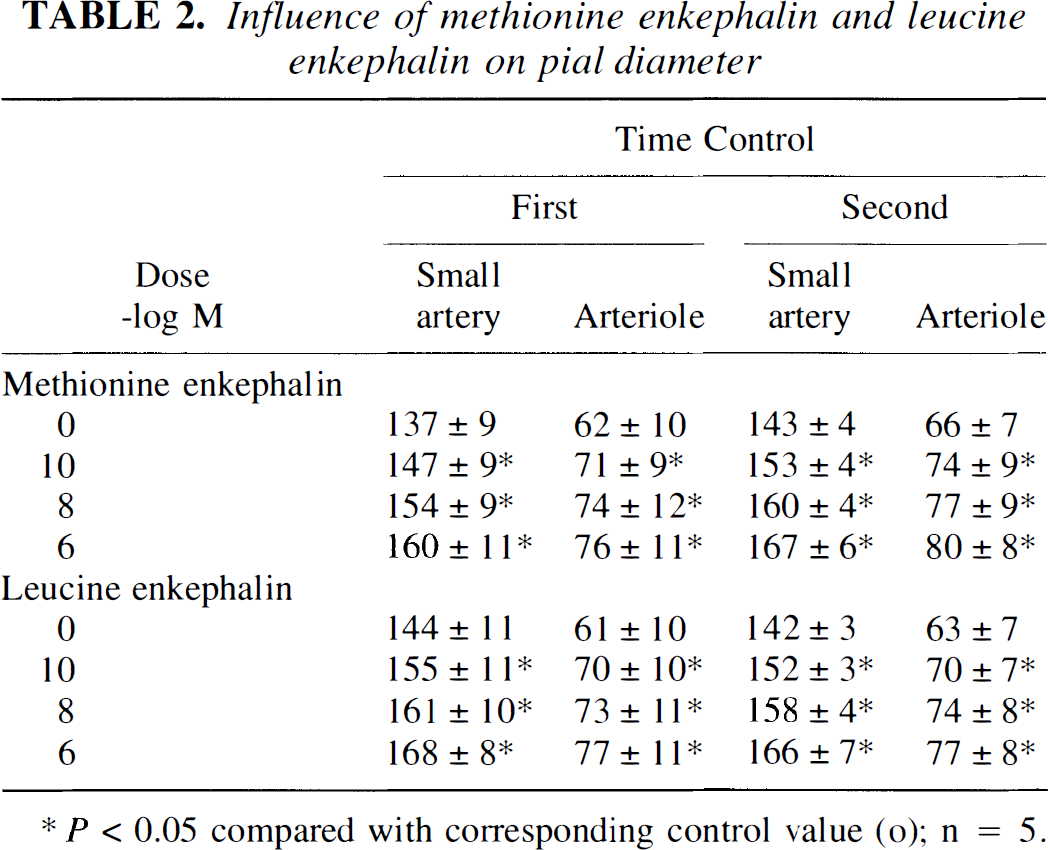
Influence of l-NIO on methionine enkephalin–induced and leucine enkephalin–induced pial artery dilation and increased cGMP concentration
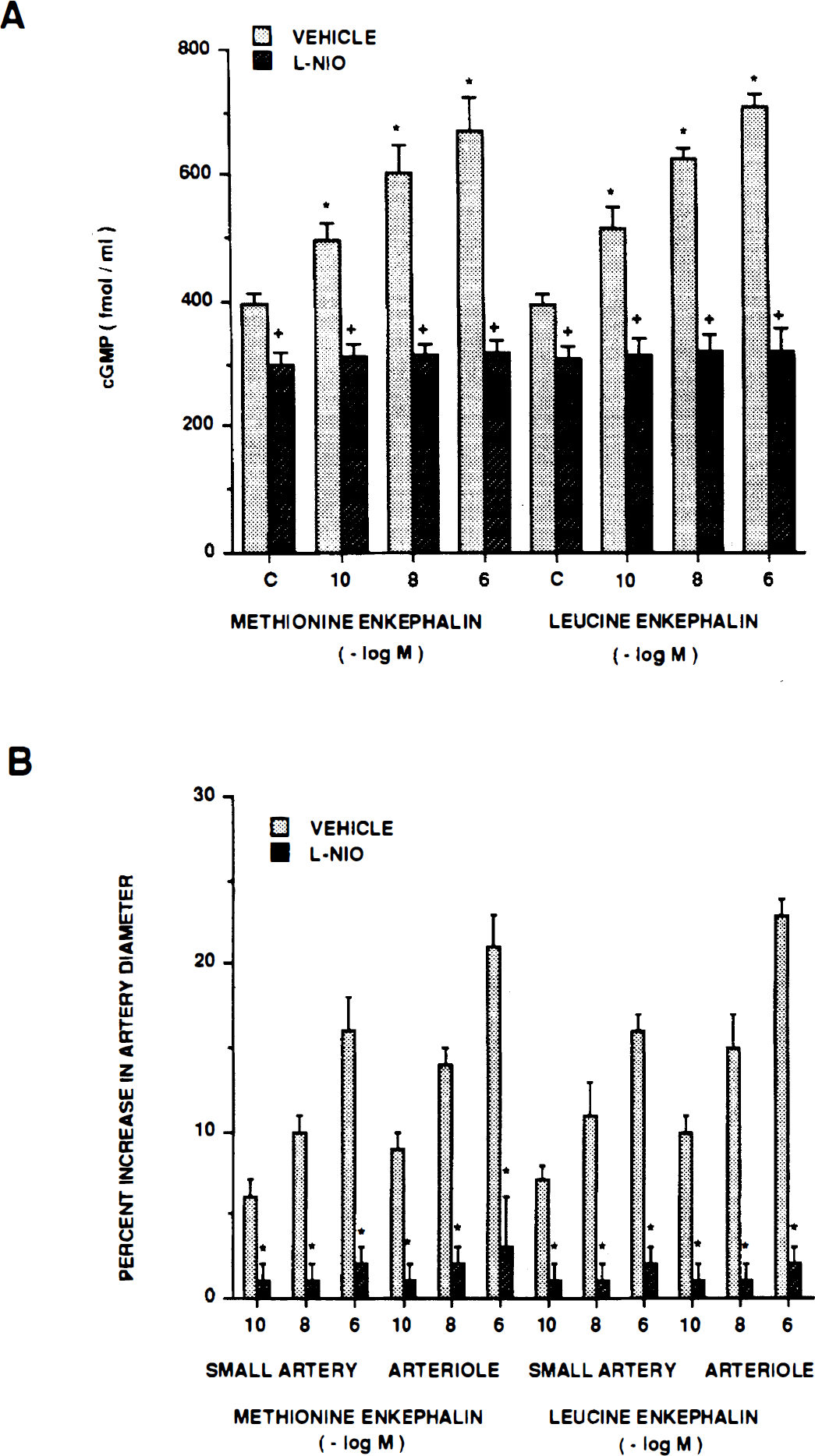
Methionine enkephalin and leucine enkephalin (10−10, 10−8, 10−6 mol/L) elicited reproducible pial small artery and arteriole vasodilation (Table 2). Dilation to both opioids was blocked by l-NIO (Fig. 3). Increases in cGMP associated with opioid dilation also were blocked in the presence of l-NIO (Fig. 3). On a fold basis, these values were 1.3 ± 0.1, 1.5 ± 0.1, and 1.7 ± 0.1 versus 1.0 ± 0.1, 1.0 ± 0.1, and 1.1 ± 0.1 for methionine enkephalin 10−10, 10−8, 10−6 mol/L, respectively, before and after coadministration of l-NIO.
(A) Influence of methionine enkephalin and leucine enkephalin (10−10, 10−8, 10−6 mol/L) on cortical periarachnoid CSF cGMP concentration (fmol/mL) in the absence (vehicle) and presence of n-iminoethyl-l-omithine (l-NIO) (10−6 mol/L). Values are mean ± SD, n = 6. *P<0.05 compared with corresponding control value (c). +P<0.05 compared with corresponding vehicle value. (B) Influence of methionine enkephalin and leucine enkephalin on pial small artery and arteriole diameter in the absence (vehicle) and presence of l-NIO (10−6 mol/L). Values are mean ± SD, n = 8. *P<0.05 compared with corresponding vehicle value.
Influence of methionine enkephalin and leucine enkephalin on pial diameter
P<0.05 compared with corresponding control value (o); n=5.
Influence of l-NIO on SNP, NMDA, and substance P–induced pial artery dilation and increased CSF cGMP concentration
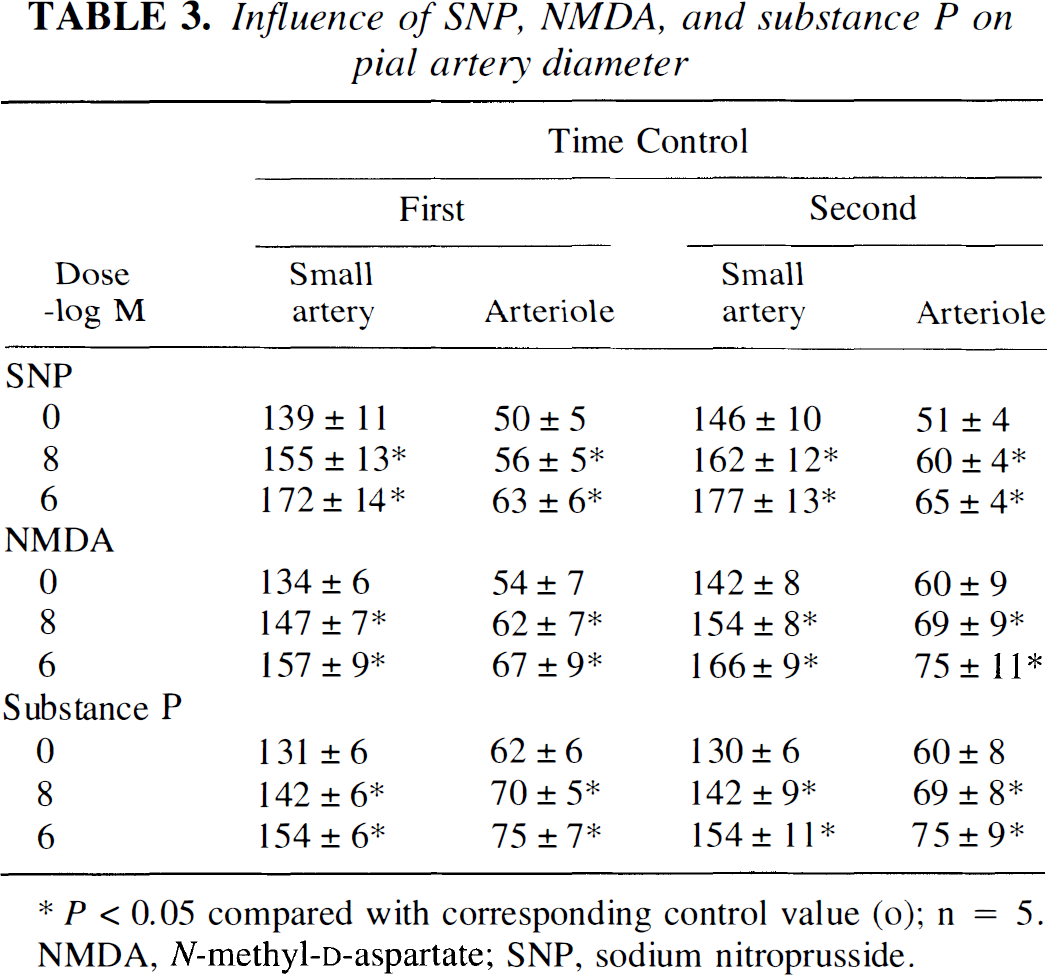
Sodium nitroprusside, NMDA, and substance P (10−8, 10−6 mol/L) elicited reproducible pial small artery and arteriole vasodilation (Table 3). The SNP- and NMDA-induced pial dilation was unchanged by l-NIO (Fig. 4). Associated increases in CSF cGMP concentrations also were unchanged in the presence of l-NIO (Fig. 4). In contrast, substance P–induced vasodilation was blunted in the presence of l-NIO (Fig. 4). Additionally, associated increases in CSF cGMP levels were blocked with the coadministration of l-NIO (Fig. 4).
(A) Influence of SNP, NMDA, and substance P (10−8, 10−6 mol/L) on cortical periarachnoid CSF cGMP concentration in the absence (vehicle) and presence of n-iminoethyl-l-omithine (l-NIO) (10−6 mol/L). Values are mean ± SD, n = 6. *P<0.05 compared with corresponding control value (c). +P<0.05 compared with corresponding vehicle value. (B) Influence of sodium nitroprusside (SNP), N-methyl-d-aspartate (NMDA), and substance P on pial small artery and arteriole diameter in the absence (vehicle) and presence of l-NIO (10−6 mol/L). Values are mean ± SD, n = 8.
Influence of SNP, NMDA, and substance P on pial artery diameter
Influence of l-NIO on pial artery diameter, CSF cGMP, and CSF opioid concentration
The l-NIO decreased resting control diameter of pial small arteries from 121 ± 12 to 112 ± 12 μm, whereas pial arterioles decreased from 60 ± 9 to 54 ± 7 μm, n = 7. The l-NIO also decreased resting control CSF cGMP concentration (Fig. 1, Fig. 3, and Fig. 4). In contrast, l-NIO had no effect on control CSF methionine enkephalin or leucine enkephalin concentration (Fig. 2).
Role of opioids in SNP-induced pial artery dilation
To determine if SNP-associated release of opioids contributes to the vascular response of that agent, responses to SNP were obtained in the absence and presence of naloxone (1 mg/kg intravenously). Pial dilation produced by SNP was unchanged by naloxone (8 ± 1 and 16 ± 2 versus 7 ± 1 and 17 ± 2% before and after naloxone, respectively, n = 5).
Blood chemistry
Blood chemistry values were obtained at the beginning and end of all normoxia experiments. These values were unchanged at the end compared with the values obtained at the beginning (7.44 ± 0.08, 33 ± 4, 99 ± 9, and 69 ± 8 versus 7.44 ± 0.08, 33 ± 5, 98 ± 9, and 65 ± 8 mm Hg for pH, Pco2, Po2, and mean arterial blood pressure, respectively, n = 34). The blood chemistry also was obtained during normoxia and hypoxia in experiments designed to investigate the cerebrovascular effects of hypoxia. Hypoxia decreased Po2 from 99 ± 12 to 35 ± 3 mm Hg (n=29) for moderate hypoxia and from 99 ± 11 to 26 ± 2 mm Hg (n = 29) for severe hypoxia, whereas pH, Pco2, and mean arterial blood pressure remained unchanged.
DISCUSSION
Results of the current study show that hypoxia-induced pial artery dilation and associated rise in cortical periarachnoid CSF cGMP concentration were unchanged after administration of l-NIO, an antagonist thought to have greater endothelial NO synthase inhibitory selectivity (Rees et al., 1990). Pial dilation and associated production of cGMP by SNP and NMDA were similarly unchanged by l-NIO. Sodium nitroprusside is well known to elicit dilation through the release of NO and not the activation of NO synthase. Alternatively, NMDA causes neuronal production and release of NO in brain tissue (Faraci and Breese, 1993; Faraci and Brian, 1995). In contrast, pial dilation and associated production of CSF cGMP by substance P was blunted by l-NIO. The observation that substance P elicits pial dilation in the newborn pig dependent on a NO but independent of a prostaglandin mechanism (Busija and Chen, 1992) was helpful in data interpretation. Taken together, then, the data in the current study showing that l-NIO had no effect on responses to SNP and NMDA but blunted pial dilation to substance P suggest that this agent preferentially inhibits endothelial over neuronal NO synthase. Additionally, recent data show that the sodium salt, 7-NINA, of the purported neuronal NO synthase inhibitor, 7-NI, blocked responses to NMDA while leaving substance P dilation unchanged, which supports this suggestion (Wilderman, 1997b and Armstead, 1997b). Responses to hypoxic pial dilation and associated production of cGMP were unchanged by l-NIO, but had previously been observed to be blunted by l-NNA (Armstead, 1995b; Wilderman and Armstead, 1996). Data in the current study accordingly indicate by exclusion that endothelial NO synthase does not contribute to dilation for that stimulus. Therefore, these data suggest that neuronal NO synthase must be the isoform used by hypoxia to elicit dilation in the piglet cerebral circulation. This suggestion is consistent with the results of a recent study using 7-NINA as a probe for the role of the neuronal isoform of NO synthase in hypoxic dilation (Wilderman, 1997b and Armstead, 1997b). However, the use of such a pharmacologic approach is indirect and heavily dependent on the selectivity of the agents used.
Additional results of the current study show that hypoxia-associated production of the opioids methionine enkephalin and leucine enkephalin also was unchanged by l-NIO. Previously, it had been observed that SNP, a releaser of NO, increased CSF methionine enkephalin and leucine enkephalin concentration, and the coadministration of LY83583 or Rp 8-Bromo cGMP (soluble guanylate cyclase and cGMP antagonists, respectively) blocked these increases in CSF opioid concentration (Wilderman and Armstead, 1996). These opioids are vasodilators (Armstead et al., 1991) and themselves have been observed to contribute to hypoxic pial dilation (Armstead, 1995a,
b
). Since hypoxic release of these opioids was attenuated by l-NNA, it had been concluded that NO-induced release of cGMP contributed to hypoxic opioid release (Wilderman and Armstead, 1996). Data from the current study, therefore, extend these observations and indicate that the activation of endothelial NO synthase does not contribute to hypoxia-associated opioid release and thus to the resulting pial artery vasodilation. Since l-NNA has been observed to blunt the hypoxic dilator and opioid release, these data suggest that the neuronal isoform of NO synthase contributes to hypoxia-associated opioid release. This suggestion is consistent with the recent observation that 7-NINA attenuated hypoxic release of CSF opioids (Wilderman 1997b and Armstead, 1997b).
In contrast to its inability to influence hypoxic pial dilation and opioid production, results from the current study showing that l-NIO blunted methionine enkephalin and leucine enkephalin pial dilation and associated CSF cGMP production indicate that activation of endothelial NO synthase does contribute to opioid-induced pial artery vasodilation. Since l-NNA has previously been observed to blunt the dilation and block associated rises in CSF cGMP concentration by these opioids (Devine and Armstead, 1995), the current study confirms and extends these findings by suggesting that it is the endothelium-derived isoform of NO synthase that contributes to these vascular and biochemical events. More importantly, because the source of NO synthase used for opioid release and vasodilation appear to be different, a feed-forward cycle linking these biochemical and vascular events does not exist.
However, the observations that l-NIO had no effect on hypoxic dilation, which is partly opioid dependent, but blunted dilation to exogenous opioids produces another conundrum. Several possibilities could explain these data. First, whereas NO and opioids do interact, these two systems can independently contribute to hypoxic pial dilation as well (Armstead, 1995b). New data in the current study showing that naloxone and l-NIO have no effect on SNP-induced pial dilation support this idea. Second, although new data in the current report show that a substantial component for the opioid dilation resulted from activation of endothelial NO synthase, other published work shows that other mechanisms (e.g., cAMP) also significantly contribute to opioid dilation (Armstead, 1997a). As described in greater detail later, hypoxic pial dilation is multifactorial. Therefore, l-NIO may blunt opioid dilation under normoxic conditions and may not have any effect on hypoxic dilation, even though opioids contribute to that dilation in that other second messengers, which presumably are unaffected by l-NIO, can mediate opioid dilation and thereby still contribute to hypoxic dilation. We speculate that mechanisms like cAMP and PACAP, which have been shown to link opioids to hypoxic dilation (Wilderman 1997a and Armstead, 1997a), can compensate for the inhibition set forth by l-NIO administration.
Finally, results of this study show that l-NIO reduced resting CSF cGMP levels and constricted pial arteries, which is consistent with the idea that endothelial NO synthase contributes to resting vascular tone (Moncada et al., 1991). However, l-NIO had no influence on resting CSF methionine enkephalin and leucine enkephalin concentration, indicating that endothelial NO synthase does not contribute to basal cerebral production of these two opioids.
Previous studies indicate that l-NIO is a selective inhibitor of endothelial NO synthase (Palacios et al., 1989; Rees et al., 1990). On the other hand, l-NIO also has been observed to inhibit NO synthase in brain tissue (Knowles et al., 1990), suggesting that its action profile may not be that selective for the endothelial isoform of NO synthase. Differences between the latter study and the current one may result from species, age, dose, or regional differences. Although supportive of a selective endothelial NO synthase interaction, the current indirect pharmacologic approach should, therefore, be interpreted with caution.
Previous studies in the newborn pig have attempted to characterize the signal transduction pathways used by NO and opioids in their contribution to hypoxic pial artery dilation. For example, pial artery vasodilation by SNP (a releaser of NO) and 8-Bromo cGMP (a cGMP analogue) was attenuated by the ATP-sensitive K+ channel (KATP) antagonist, glibenclamide (Armstead, 1996c), whereas hypoxia-induced, methionine enkephalin–induced, and leucine enkephalin–induced pial dilation all were similarly attenuated by this antagonist (Shankar and Armstead, 1995). It has been shown that NO primarily elicits its vascular effects via cGMP production in the piglet circulation because LY83583 and Rp 8-Bromo cGMP, soluble guanylate cyclase and protein kinase G inhibitors, respectively, blunted SNP-induced vasodilation (Armstead, 1996c; Wilderman 1996) and Armstead, 1996. In contrast, responses to SNP and 8-Bromo cGMP were unchanged in the presence of the calcium sensitive K+ channel antagonist, iberiotoxin (Armstead, 1997b). Therefore, these data indicate that endothelial NO-induced production of cGMP results in the activation of the KATP channel and pial artery dilation due to the opioids methionine enkephalin and leucine enkephalin. Differences in study design could account for the recent opposing observation that SNP elicits dilation independent of KATP channel activation (Bari et al., 1996).
Because l-NIO did not alter hypoxic pial dilation but the light/dye endothelial injury technique did (Leffler et al., 1997), these data suggest that endothelial products other than NO contribute to the vascular response to this stimulus. As suggested by Leffler et al. (1997), cytochrome P450 epoxygenase metabolites of arachidonic acid could be one example of such an endothelial product.
Other published data indicate that additional mechanisms contribute to hypoxic pial artery dilation in the newborn pig. For example, it has recently been observed that cAMP contributes to both hypoxic pial dilation and associated opioid release in the piglet (Wilderman, 1997a and Armstead, 1997a). Similarly, prostaglandins and vasopressin also contribute to hypoxic dilation and associated opioid release via both cGMP and cAMP-dependent mechanisms (Armstead, 1996a,
b
; Rossberg and Armstead, 1996, 1997). In contrast, adenosine contributed to hypoxic pial dilation via NO, cyclic nucleotide and KATP channel–dependent mechanisms independent of the release of opioids (Armstead, 1997c). Since in all of the above studies pharmacologic probes for each vasoactive system could attenuate but not completely eliminate hypoxic pial dilation, these data taken together indicate that dilation to this stimulus is multifactorial. It is speculated that as one vasoactive system is eliminated, others are upregulated to buffer the removal of the contributing mechanism. Reasons for differences between our previously published data indicating a role for NO in hypoxic pial dilation and those of Leffler et al (1997) which do not are uncertain, but could relate to a more prolonged hypoxic exposure in our studies.
Potential sources of opioids and cyclic nucleotides in cortical periarachnoid CSF are neurons, glia, vascular smooth muscle, and endothelial cells. However, the source of these substances cannot be determined from the current experimental design.
In conclusion, results of the current study show that endothelial NO synthase contributes to opioid-induced pial artery dilation but not hypoxia-induced dilation. Additionally, these data suggest that neuronally derived NO contributes to hypoxic pial dilation.
Footnotes
Acknowledgments
The authors thank Joseph Quinn for his exceptional technical assistance when performing the experiments.
Abbreviations used
References
1.
ArmsteadWM (1997a) Role of activation of calcium sensitive K+ channels and cAMP in opioid-induced pial artery dilation. Brain Res747:252–258
2.
ArmsteadWM (1997b) Role of activation of calcium sensitive K+ channels in nitric oxide and hypoxia induced pial artery dilation. Am J Physiol272:H1785–H1790
3.
ArmsteadWM (1997c) Role of nitric oxide, cyclic nucleotides, and the activation of ATP sensitive K+ channels in the contribution of adenosine to hypoxia-induced pial artery dilation. J Cereb Blood Flow Metab17:100–108
4.
ArmsteadWM (1996a) cGMP and cAMP in prostaglandin induced pial artery dilation and increased CSF opioid concentration. Am J Physiol271:H166–H172
5.
ArmsteadWM (1996b) Relationship between opioids and prostaglandins in hypoxia-induced vasodilation of pial arteries in the newborn pig. Proc Soc Exp Biol Med212:135–141
6.
ArmsteadWM (1996c) Role of ATP sensitive K+ channels in cGMP-mediated pial artery vasodilation. Am J Physiol270:H423–H426
7.
ArmsteadWM (1995a) The contribution of delta 1 and delta 2 opioid receptors to hypoxia induced pial artery dilation in the newborn pig. J Cereb Blood Flow Metab15:539–546
8.
ArmsteadWM (1995b) Opioids and nitric oxide contribute to hypoxia induced pial artery vasodilation. Am J Physiol268(Heart Circ Physiol):H226–H232
9.
ArmsteadWMMirroRBusijaDWLefflerCW (1991) Opioids in cerebrospinal fluid in hypotensive newborn pigs. Circ Res68:922–929
10.
BariFErricoRALouisTMBusijaDW (1996) Interaction between ATP-sensitive K+ channels and nitric oxide on pial arterioles in piglets. J Cereb Blood Flow Metab16:1158–1164
11.
BusijaDWChenJ (1992) Effects of trigeminal neurotransmitters on piglet pial arterioles. J Develop Phys18:67–72
12.
CoyleMGOhWStonestreetBS (1993) Effects of indomethiacin on brain blood flow and cerebral metabolism in hypoxic newborn piglets. Am J Physiol264:H141–H149
13.
DevineJArmsteadWM (1995) The role of nitric oxide in opioid induced pial artery vasodilation. Brain Res675:257–263
14.
EisenachJCTongCStumpDABlockSM (1992) Vasopressin and fetal cerebrovascular regulation. Am J Physiol263:R376–R381
15.
EmersonTBJrRaymondRM (1981) Involvement of adenosine in cerebral hypoxia hyperemia in the dog. Am J Physiol241:H134–H138
16.
FaraciFMBreeseKR (1993) Nitric oxide mediates vasodilation in response to activation of N-methyl-d-aspartate receptors in brain. Circ Res72:476–480
17.
FaraciFMBrianJEJr (1995) 7-Nitroindazole inhibits brain nitric oxide synthase and cerebral vasodilation in response to N-methyl-d-aspartate. Stroke26:2172–2176
KellyPATRitchieIMArbuthnottGW (1995) Inhibition of neuronal nitric oxide synthase by 7-nitroindazole: effects upon local cerebral blood flow and glucose use in the rat. J Cereb Blood Flow Metab15:766–773
22.
KnowlesRGPalaciosMPalmerRMJMoncadaS (1990) Kinetic characteristics of nitric oxide synthase from rat brain. Biochem J269:207–210
23.
KozniewskaEOsakaMStysT (1992) Effect of endothelium derived nitric oxide on cerebral circulation during normoxia and hypoxia in the rat. J Cereb Blood Flow Metab12:311–317
24.
LefflerCWSmithJSEdringtonJLZuckermanSLParfenovaH (1997) Mechanisms of hypoxia-induced cerebrovascular dilation in the newborn pig. Am J Physiol272:H1323–H1332
25.
MartinezAMPadburyJFBurnellEFThioSL (1991) Plasma methionine enkephalin levels in the human newborn at birth. Biol Neonate60:102–103
26.
MartinezAMPadburyJFBurnellEFThioSLHummeJ (1990) The effects of hypoxia on methionine enkephalin peptide and catecholamine release in fetal sheep. Pediatr Res27:52–55
27.
MoncadaSPalmerRMJHiggsEA (1991) Nitric Oxide: Physiology, pathophysiology, and pharmacology. Pharm Rev43:109–142
28.
PalaciosMKnowlesRGPalmerRMJMoncadaS (1989) Nitric oxide from l-arginine stimulates the soluble guanylate cyclase in adrenal glands. Biochem Biophys Res Commun165:802–809
29.
PelligrinoDAWangQKoenigHMAlbrechtRF (1995) Role of nitric oxide, adenosine, N-methyl-d-asparatate receptors, and neuronal activation in hypoxia-induced pial arteriolar dilation in rats. Brain Res704:61–70
30.
ReeseDDPalmerRMJSchultzRHodsonHFMoncadaS (1990) Characterization of three inhibitors of endothelial nitric oxide synthase in vitro and in vivo. Br J Pharmacol101:746–752
31.
RossbergMIArmsteadWM (1997) Role of cyclic nucleotides in vasopressin-induced piglet pial artery dilation and opioid release. Pediatric Res41:498–504
32.
RossbergMIArmsteadWM (1996) Relationship between vasopressin and opioids in hypoixia induced pial artery vasodilation. Am J Physiol271:H521–H527
33.
SankaranKHindmarshKVWatsonVG (1983) Hypoxic-ischemic encephalopathy and plasma Beta endorphin. Dev Pharmacol Ther7:377–383
34.
ShankarVArmsteadWM (1995) Opioids contribute to hypoxia induced pial artery dilation through the activation of ATP-sensitive potassium channels. Am J Physiol269:H998–H1002
35.
WangQPelligrinoDABaughmanVLKoenigHMAlbrechtRF (1995) The role of neuronal nitric oxide synthase in regulation of cerebral blood flow in normocapnia and hypercapnia in rats. J Cereb Blood Flow Metab15:774–778
36.
WildermanMJArmsteadWM (1996) Relationship between nitric oxide and opioids in hypoxia-induced pial artery vasodilation. Am J Physiol270:H869–H874
37.
WildermanMJArmsteadWM (1997a) Role of PACAP in the relationship between cAMP and opioids in hypoxia-induced pial artery vasodilation. Am J Physiol272:H1350–H1358
38.
WildermanMJArmsteadWM (1997b) Role of neuronal nitric oxide synthase in the relationship between nitric oxide and opioids in hypoxia induced pial artery dilation. Am J Physiol273:H1807–H1815