
Abstract
Elevation of blood flow velocity in the large cerebral vessels is known to be of substantial pathophysiologic and prognostic significance in sickle-cell disease (SCD). Its precise cause is not established, but the two obvious proximal mechanisms are obstructive vascular stenosis and hemodynamic dilatation. Here we revisit this distinction by analyzing cerebrovascular reserve capacity. Forty-two patients with SCD underwent measurements of global cerebral blood flow in grey matter by the 133Xe inhalation method during normocapnia and hypercapnia to quantify cerebrovascular reactivity. Cerebral blood flow was significantly higher in SCD patients (120±31 ml/100 g/min) than in controls (76±20 ml/100 g/min). Reactivity was significantly lower in SCD patients (1.06±1.92 versus 2.16±1.15%/mm Hg). Stepwise multiple regressions within the SCD sample determined that normocapnic cerebral blood flow was largely predicted by hematocrit (r =–0.59; P > 0.0001), whereas hypercapnic reactivity was only predicted by normocapnic flow across all subjects (r =–0.52; P > 0.0001). None of the controls, but 24% of the SCD patients showed ‘steal’ (negative reactivity, χ2 = 6.05; P > 0.02). This impairment of vasodilatory capacity, occurring at perfusion levels above 150 ml/100 g/min, may reflect intrinsic limitations of the human cerebrovascular system and can explain both the elevated blood flow velocities and the high risk of stroke observed in such patients.
Keywords
Introduction
One of the major consequences of sickle-cell disease (SCD) is a high risk of stroke: SCD is the most common cause of stroke in children. More than 10% of SCD patients will have a clinical first stroke by age 20, and another 20 to 30% will have a silent infarction (Switzer et al, 2006). The pathophysiology of stroke in SCD has been a subject of intense research for decades, and it is known to involve several interacting mechanisms at the molecular and vascular levels (Kirkham and Datta, 2006; Prengler et al, 2002).
Despite the predisposition to distal insufficiency infarction, indicating ischemia (Pavlakis et al, 1988), we documented severe hyperemia in these patients (Prohovnik et al, 1989). The mechanism relating cerebral hyperemia to ischemic infarcts was unknown at the time. Later, Adams and colleagues reported that elevated arterial blood flow velocities measured by transcranial Doppler (TCD) predicted stroke risk in SCD patients. Although this observation was consistent with cerebral hyperemia, its relationship to the occurrence of ischemic stroke was obscure. Initially, it was assumed that the high velocity was related to high-grade large vessel stenosis, but the majority of patients had normal MRA in the presence of high TCD velocities (Abboud et al, 2004). Nevertheless, many authors continue to interpret high TCD velocities as a manifestation of large-vessel arteriopathy, despite evidence inconsistent with this interpretation. For example, Bernaudin et al (2005) found that hydroxyurea treatment raised Hb and hematocrit (Hct), and then led to decreased TCD velocity; this is unlikely to be due to amelioration of stenosis.
Here we present cerebral blood flow data that can explain both the elevated blood flow velocities and the ischemic strokes in SCD, without the need to postulate a high prevalence of major vessel stenosis (which is not supported by the evidence). This hemodynamic model of ischemic stroke may have therapeutic ramifications in SCD, and possibly other stroke syndromes.
Materials and methods
This paper presents our experience measuring cerebrovascular reactivity in 42 consecutive SCD patients. Where patients were followed over time, we included only their first observation that had technically satisfactory reactivity data. Baseline data (without reactivity measures) of the current patients were part of a previous publication (Prohovnik et al, 1989), which also included detailed characteristics of the patient population (88% of the patients had hemoglobin SS). We now present their cerebral reactivity data in a regression analysis; for illustration purposes, we also include 13 nonhematologic young patients of similar age and gender distribution. The study was approved by the IRB, and consents were signed by adult subjects and the parents of minors.
To measure vasodilatory capacity of cerebral vasculature, we used the standard CO2 challenge method. Cerebral blood flow (CBF) was quantified by the 133Xe inhalation method (Novo Cerebrograph 32c, Meditronic, Hadsund, Denmark). Measurements were made at normocapnia (breathing room air) and hypercapnia (breathing air enriched with 4% CO2) (Gitelman et al, 1991). All data were evaluated and analyzed using the six-unknown model (Prohovnik et al, 1983) with standard quality control measures (Prohovnik, 1988). Flow is presented as fg, for the grey-matter compartment, and is averaged over the 32 regions. Baseline systolic, diastolic, and mean blood pressure and pulse were taken immediately after the normocapnic run was completed; during the hypercapnic state, vital signs were taken approximately every 2 to 3 min throughout the run, and the data set with the highest systolic pressure reading was used in the current analysis. End-tidal CO2 (ETpCO2) was monitored continuously during each run by means of a capnograph hooked to the breathing circuit. We employed ETpCO2 as an estimate of PaCO2 given our exclusion of patients with severe chronic obstructive pulmonary disease; we have shown elsewhere a good agreement between the two during general anesthesia (Young et al, 1991). We computed the customary percentage reactivity (Δ% fg/Δ ETpCO2 = Δ fg/resting fg/Δ ETpCO2), where Δ indicates a change (difference) and Δ% indicates change in percentage units.
Statistical analyses included linear and nonlinear regressions, stepwise multiple regressions and appropriate analysis of covariance models. All analyses were performed by JMP software (SAS Institute Inc, Cary, NC) running on the Macintosh OS X operating system, using 0.05 for nominal significance threshold.
Results
Sample characteristics and physiologic variables are detailed in Table 1. The SCD sample is younger, because it includes pediatric patients (age range 8 to 32 years), whereas the controls were all adult (age range 19 to 35 years). There were no significant differences between the groups in either normocapnic or hypercapnic pCO2, or in their difference, indicating equivalent pulmonary function.
Sample characteristics
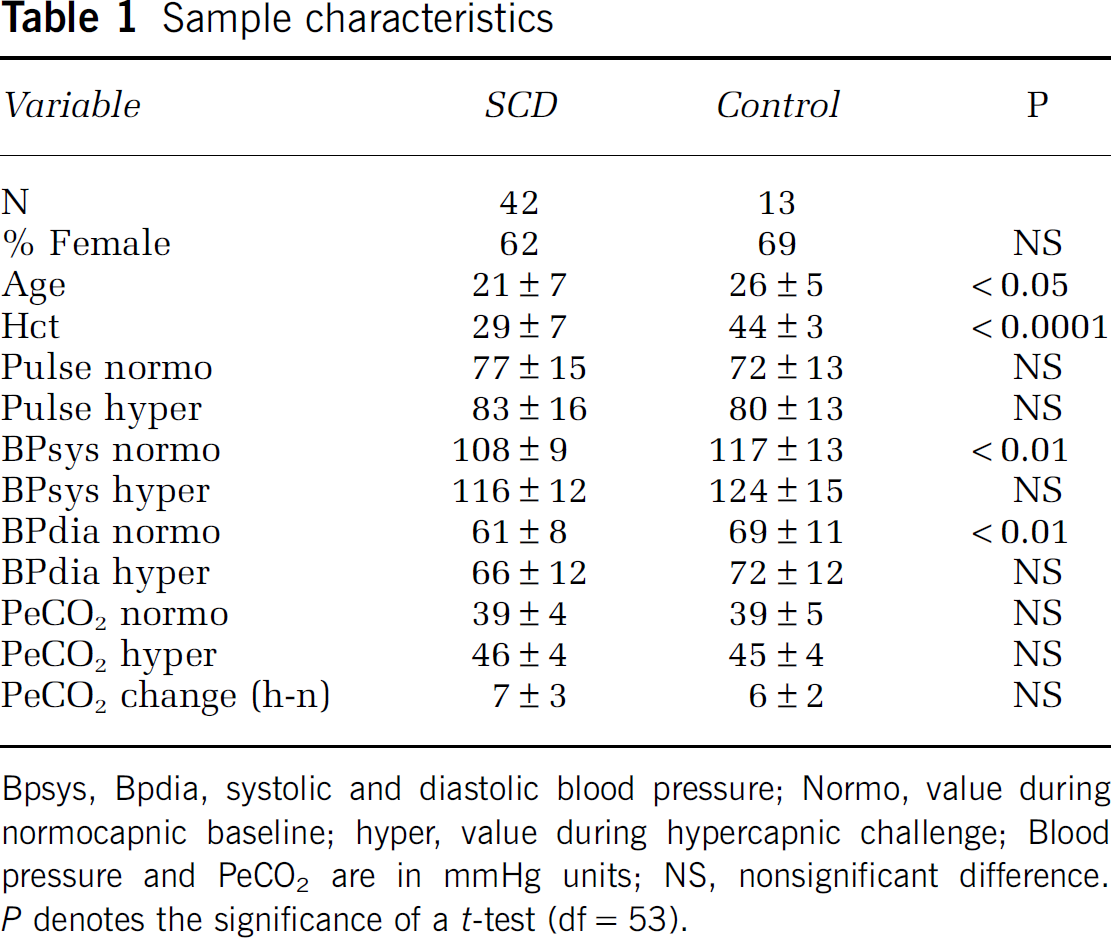
Bpsys, Bpdia, systolic and diastolic blood pressure; Normo, value during normocapnic baseline; hyper, value during hypercapnic challenge; Blood pressure and PeCO2 are in mmHg units; NS, nonsignificant difference. P denotes the significance of a t-test (df=53).
Normocapnic Cerebral Blood Flow
Cerebral blood flow was higher in SCD (120±31) than in the controls (76±20 ml/100 g/min), as expected (t53 = 4.88; P > 0.0001). Analysis of covariance on normocapnic flow, using diagnosis as between-group factor and age and Hct as covariates yielded no significant diagnosis effect, but highly significant effects for Hct (F1,51 = 37.34; P > 0.0001) and age (F1,51 = 19.81; P > 0.0001), as expected.
Across all subjects (n = 55), CBF was significantly correlated with age (r =–0.44; P = 0.0009; CBF = 155–2.08 × age) and even more strongly with Hct (r =–0.71; P > 0.0001; CBF = 196–2.68 × Hct). A stepwise multiple regression was used to search for other predictors of cerebral perfusion among all physiologic observations (pulse rate, blood pressure, pCO2) but found only Hct and age to be significant predictors (P > 0.0001), and a multiple regression using both yielded r =–0.80 (P > 0.0001). Repeating the regressions within each subsample showed no significant age or Hct correlation within the control sample. Within the SCD patients, age yielded a significant correlation (CBF = 151–1.50 × age; r =–0.35; P > 0.05), but Hct was clearly the best predictor (CBF = 194–2.59 × Hct; r =–0.59; P > 0.0001; Figure 1).
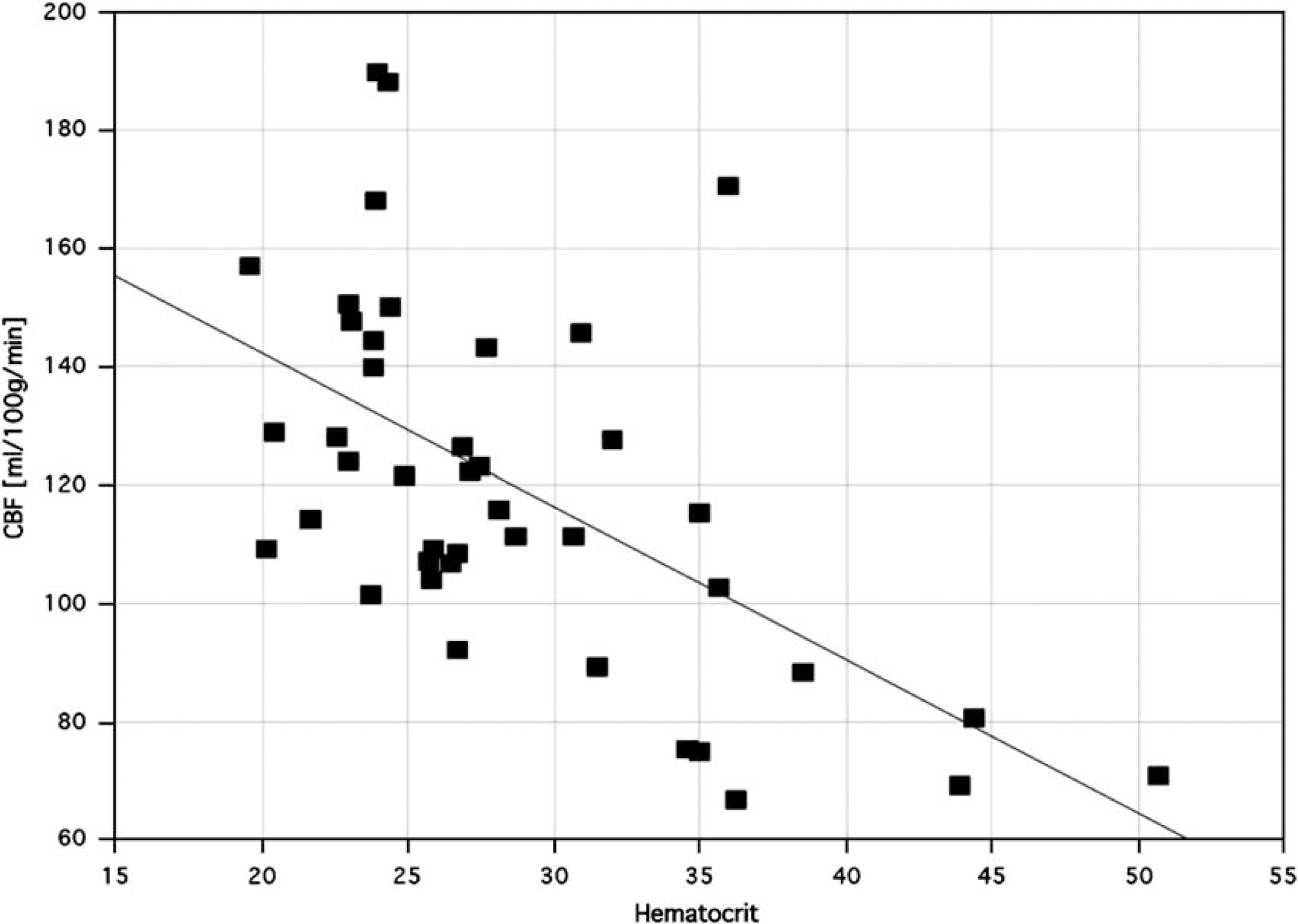
Baseline CBF as a function of hematocrit in SCD pateints. The continuous line depicts the linear regression.
Hypercapnic Reactivity
Reactivity was substantially higher in controls, more than double the value in SCD patients (2.15% versus 1.06%), but the significance was marginal (t53 = 1.94; P > 0.06) due to high variability within the patients. The reason for this high variability, and a much higher significance, were observed in a following analysis of covariance with diagnosis as between-subject factor and age, Hct and normocapnic flow as covariates. This analysis yielded no significant effects for diagnosis or Hct, but showed strong significance for baseline normocapnic blood flow (F1,50=18.29; P > 0.0001) and a small significant effect for age (F1,50=4.05; P > 0.05). The age effect was due to one outlier with the lowest reactivity, and disappeared when this subject was excluded. No significant correlations between age and reactivity were observed in any sample, with or without this subject.
The linear relationship between normocapnic resting flow and reactivity across all subjects (r =–0.52; P > 0.0001) is depicted in Figure 2. The linear regression equation was reactivity = 4.37–0.028 × CBF. A second-degree polynomial function fitted slightly better (r =–0.59; P > 0.0001), and both suggested a rapid decline of reactivity into negative values starting at a flow level of approximately 150 to 160 ml/100 g/min.
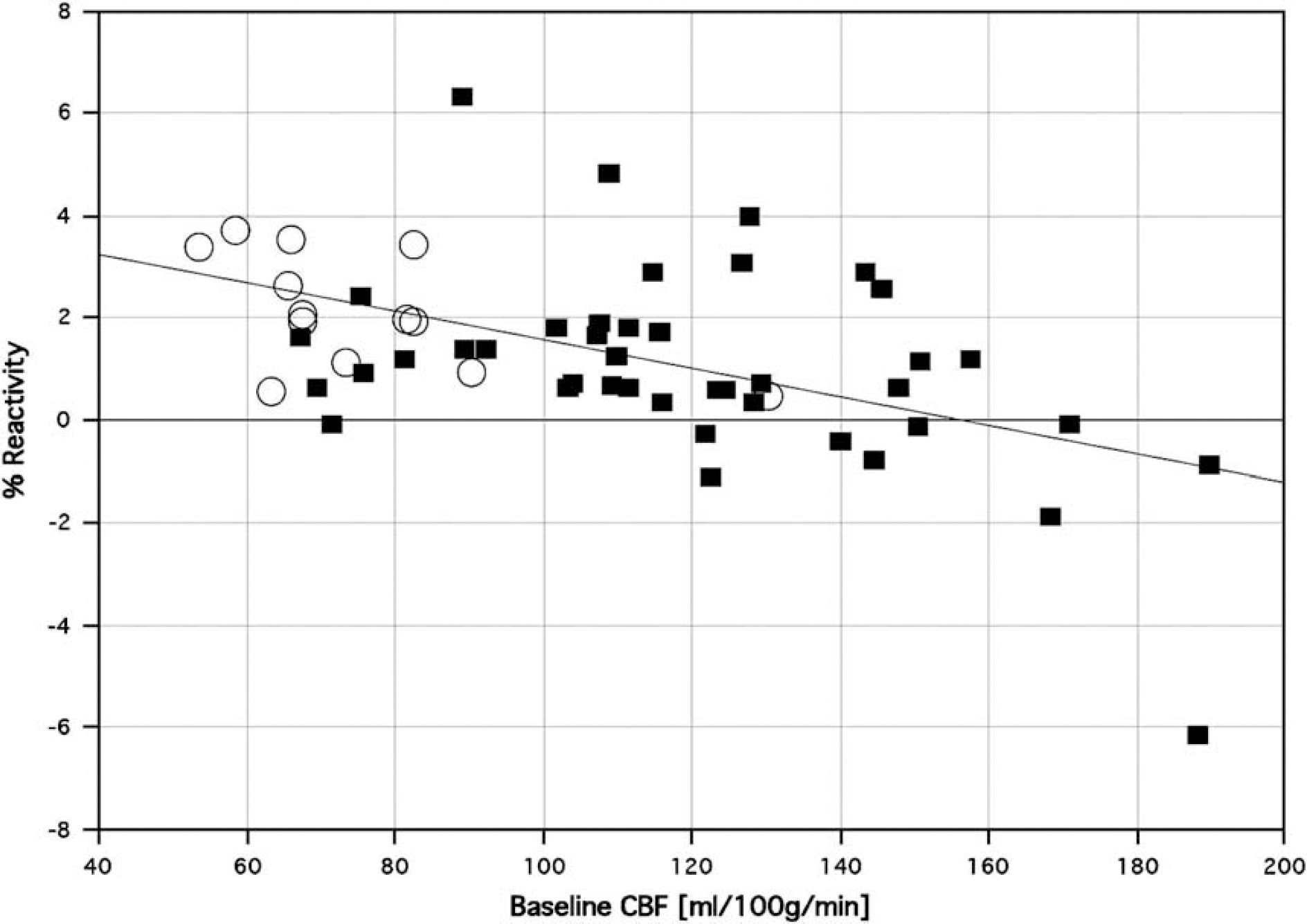
Hypercapnic reactivity against its best predictor, normocapnic blood flow. SCD patients (filled squares) and controls (empty circles) seem to fall on an identical regression line, which is linear up to about blood flow levels of 150 ml per 100 g/min but drops precipitously after that.
Discussion
Cerebral hyperemia in SCD has been amply documented previously, by several measurement methods (Brass et al, 1991; Oguz et al, 2003; Strouse et al, 2008), and is known to be anemia associated. We originally showed a linear relationship between normocapnic CBF and Hct (Prohovnik et al, 1989): within the nontransfused sample, the equation was fg = 231–3.88 × Hct. We have also calculated this regression for TCD velocity (Brass et al, 1987): transforming the reported regression to a linear one in the range of 20 to 40 of Hct yields velocity = 245–4.05 × Hct. These equations are similar to the corresponding one reported here, although the current values are lower, presumably because the sample includes older and transfused patients (fg = 194–2.59 × Hct). In adult patients, both intercept and slope are lower, but the regression equally robust, as also reported by others (Valadi et al, 2006). Thus, it is clear that cerebral perfusion in SCD patients is strongly related to the degree of anemia. Further, the relationship appears linear within the range of Hct studied, approximately 20 to 45%. This relationship is not observed in nonanemic controls.
In contrast, cerebral vasodilatory capacity, as measured by reactivity to CO2 challenge, appears to be determined by baseline blood flow, the relationship seems identical for both SCD patients and nonanemic controls, and its linearity appears to break down at blood flow levels higher than 150 ml/100 g/min. Above 160 ml/100 g/min, all subjects showed negative reactivity. Nonanemic patients usually do not reach this level of hyperemia. Finally, we have never observed, nor has anyone else to our knowledge, blood flow levels > 200 ml/100 g/min. Below we discuss the possible significance of these findings to stroke pathophysiology, prediction and treatment in this population.
Stenosis Versus Hyperemia in Sickle-Cell Disease
The STOP clinical trial of transfusion in SCD was prematurely terminated due to dramatic success: it showed that in hyperemic pediatric SCD patients, transfusion reduced stroke risk by 90% (Adams et al, 1998). In this trial, and subsequent practice guidelines, hyperemia is defined as blood flow velocity > 200 cm/s in the MCA or distal ICA. However, velocity measurements by TCD cannot determine true cerebral hyperemia, because elevated velocity may reflect hyperemia (i.e., truly higher tissue perfusion, or volume blood flow), or stenosis (increased velocity, without increased volume flow), or both. The relationship between the two, and their relative dominance in determining prognosis and treatment indications, is unknown.
Many investigators assert that the high velocities observed in SCD are due to major vessel obstructive disease, but we find the evidence weak. Early studies (Russell et al, 1984) reported a very high prevalence of obstructive cerebrovascular disease in the most severely affected SCD patients. These studies were not methodologically optimal, and modern observations suggest a much lower prevalence, and the strong possibility that major vessel stenosis may represent a consequence, rather than the cause, of stroke pathophysiology in SCD. For example, Kwiatkowski et al (2006) stated that in children ‘the majority of strokes are due to an occlusive vasculopathy affecting most commonly the distal ICA, MCA, and ACA,‘ whereas in their own data (Kwiatkowski et al, 2004), the topographic relationship between MRA abnormalities, elevated TCD velocities, and the infarcted vascular supply territory is weak. Bernaudin et al (2005) also found variable relationship between the site of MRA abnormality and elevated TCD velocities, and their data show that high TCD velocities are actually more strongly related to anemia than to MRA abnormalities. Similarly, Minniti et al (2004) suggested that transfusion normalized TCD velocities in patients with normal MRA, and thus, presumably reduced stroke risk by changing hemodynamics. Most recently, hyperemia has been confirmed in SCD patients in the absence of significant stenoses, as well as significant correlations between the level of hyperemia and cognitive deficits (Strouse et al, 2008).
Further, the STOP TCD criteria were developed in pediatric patients; in adults, despite preserved correlation of TCD velocity with Hct, these criteria do not seem to apply, as adults show much lower velocities despite continued stroke risk (Valadi et al, 2006), suggesting greater influence of hemodynamics than stenosis. Even in the STOP trial, fully 79% of the MRAs among pediatric patients with abnormally high TCD velocities who were randomized were read as normal or only mildly (<25%) stenotic (Abboud et al, 2004). Further, on extended follow-up of the STOP trial patients (Lee et al, 2006; Quinn et al, 2007), all six patients who developed stroke had elevated TCD velocities, but only two had abnormal MRAs. Of the nontransfused group, 25 patients remained stroke free and 14 did develop stroke: the only difference between these two groups was TCD velocities, higher in the stroke group. Finally, we have shown by serial observations that cerebral perfusion and TCD velocities are strongly and negatively correlated with Hct in each individual patient, and can rise or fall very quickly, within months on changing transfusion regimens (Hurlet-Jensen et al, 1994) or even within minutes during transfusion (Venketasubramanian et al, 1994). These rapid changes are inconsistent with the relatively slow rate of development of stenotic disease, but compatible with a hemodynamic etiology.
Therefore, it is unlikely that severe stenosis is the sole, or even major, pathophysiologic basis for high TCD velocities in SCD. By the same line of reasoning, if only statistically, obstructive cerebrovascular disease cannot explain the majority of ischemic infarcts among pediatric SCD patients. Stenosis of major cerebral vessels, although certainly present in SCD, cannot explain a substantial part of stroke risk, and may even be the result, rather than the cause, of high velocities (Ausavarungnirun et al, 2006). In a large proportion of SCD patients, especially during the most stroke-prone years of childhood, there is substantial hyperemia, which is tightly determined by the degree of anemia. Regardless of the presence, severity, and location of stenosis, this hyperemia would be visible to Doppler procedures as high velocity. The very high velocity used as a criterion for prophylactic transfusion in SCD is, therefore, an inevitable consequence of, and a reliable indicator of, severe anemia.
Anemia, however, exists in other conditions, where the stroke risk is much lower, such as thalassemia; therefore, there are additional risk factors in SCD. Full discussion of such factors is beyond the scope of this article, but occlusive vasculopathy is probably one. It is even possible, though we are not aware of any such evidence, that stenosis may be transient (i.e., spasm), and thus, not easy to detect by routine angiograms. Other factors, such as HbS (Hurlet-Jensen et al, 1994), hemolytic rate (O'Driscoll et al, 2008), absence of alpha thalassemia, and G6PD deficiency (Bernaudin et al, 2008), as well as NO regulation (Prengler et al, 2002), may exert an independent effect on CBF in SCD and constitutes an additional risk factor for stroke in this population. Our current data do not directly invalidate the stenosis hypothesis, as we did not obtain angiograms on these patients, but the theoretical reasoning and literature review above do not support stenosis as the primary risk factor for stroke in this population. Our current data do strongly support a major role for hyperemia in limiting cerebrovascular reserve, as detailed below.
Cerebrovascular Reserve in Sickle-Cell Disease
As showed by the STOP trial, anemia-associated hyperemia, and its expression as elevated TCD velocities, is a powerful predictor of stroke in SCD. Our current data show that this hyperemia is associated with exhaustion of cerebrovascular reserve capacity, as reflected in the vasodilatory response to hypercapnia. Not only did SCD patients show severely depleted vascular reserve, but also 24% of patients showed steal, that is, paradoxical decreases of perfusion in response to hypercapnia. For the purpose of this discussion, steal is defined operationally as negative reactivity, rather than its rigorous definition (Langer et al, 2006). Steal is not seen in healthy subjects and is rare in most pathologic populations. For example, Xie et al (2005) studied 17 patients with stable congestive heart failure with TCD; the mean hypercapnic reactivity was approximately 3%/mmHg, and none showed reactivity below 1%/mmHg. In contrast, steal was reported in the MCA cortical territory ipsilateral to an ICA or MCA occlusion in 11 of 22 patients (Okazawa et al, 2003). We (Tatemichi et al, 1988) showed in young patients (age 32±8 years) with Moyamoya disease much lower mean reactivity (0.79±0.59%/mmHg) than in other patients with occlusive CVD, including bilateral ICA occlusion. The results suggested that low reactivity might not be an indication of occlusive disease, but of the efficiency of collateral supply and vascular reserve. Notably, Moyamoya pathology is commonly found in SCD patients with a history of stroke (Switzer et al, 2006), suggesting that inefficient collateral circulation as a common hemodynamic feature.
Grave reduction of dilatory capacity can be caused by several mechanisms, one of which implicates approaching maximal dilatation. Vasodilatation is the first and most powerful compensatory response to cerebral ischemia. Flow is proportional to the fourth power of the vascular radius, so a 35% increase of diameter should increase flow rates by approximately 230%, and a 45% increase of diameter would induce a 340% flow enhancement, all else being equal. Because of this powerful relationship, even small vasodilatory changes can have major effects on cerebral perfusion. A vessel cannot dilate beyond its maximum tolerated diameter, dictated by mechanical or biochemical constraints. These limits are not yet established. The absolute mechanical limit of distensibility is probably around a doubling of diameter, as shown in pial arterioles in the rat after total muscular deactivation with EDTA (Baumbach et al, 1988). More realistically, highest neurogenic elevation of blood flow is probably reached during epileptic seizures and artificial stimulation experiments. Electrical Nucleus Basalis stimulation in the conscious rat raised cortical CBF by 150 to 350% (Vaucher et al, 1997). Potassium-induced spreading cortical depression increased CBF by 300 to 450% (Wolf et al, 1997). Therefore, under these artificial conditions, dilatory limits probably correspond to approximately 300 to 400% flow enhancement, or approximately 40 to 50% increase of diameter, but the normal physiologic range is much lower. In rats during hemorrhagic hypotension, maximal dilatation of the basilar artery (at mean blood pressure 30mmHg) was approximately 29%, whereas the smaller branching arterioles increased their diameter by approximately 44% (Toyoda et al, 1996). In rat capillaries, RBC velocity increased by 115% when Hct dropped from 45 to 17 (Hudetz et al, 2000). In adult humans, mean cerebral tissue perfusion is approximately 50 ml/100 g/min, and grey-matter flow is approximately 75 ml/100 g/min. Tripling these values would then yield approximately 215 ml/100 g/min for grey matter as the upper limit of flow rates during physiologic conditions. Our flow values in SCD are also in good agreement with actual dilatation measurements of the basilar artery (Steen et al, 1998), documented at 23% dilation of the basilar artery in SCD (4.3±0.6 versus 3.5±0.7), inversely correlated with Hct and WAIS.
Therefore, it appears that cerebral vessels do not dilate, within physiologic range, by more than approximately 30 to 40% (in diameter), corresponding to triple the baseline perfusion rate. In humans, the limit would be approximately 215 ml/100 g/min in grey matter. Our current data show that cerebrovascular hypercapnic reactivity is strongly predicted by baseline perfusion, and it decreases rapidly and disappears in grey matter at values of 160 to 200 ml/100 g/min. These data are in good agreement with the maximal dilatation hypothesis.
Pathophysiologic Implications
Our data show that the anemia-associated cerebral hyperemia of SCD is a likely cause of the TCD-measured high velocities. This hyperemia is especially high in children, where the TCD velocity cutoff value of 200 cm/sec has been found to predict stroke in the STOP trial. Cerebral vessels may also be slightly less distensible in children, as suggested by smooth muscle deactivation by EDTA in 3- to 4-month-old rats (Baumbach et al, 1988). The association of hyperemia with stroke is also consistent with their known age distributions: both stroke incidence and cerebral blood flow (and large vessel velocities) peak around age 5 to 9 years, and then decrease continuously toward early adulthood (Ausavarungnirun et al, 2006).
Thus, our data, as well as previous work, can be parsimoniously interpreted by the following model. Baseline cerebral perfusion in SCD is mostly determined by age and the severity of anemia. Cerebral blood flow would, therefore, be highest in young children with the worst anemia, where it will also be manifested in very high TCD velocities. Under physiologic conditions, the upper limit of cerebral dilatation corresponds to a tissue perfusion rate of approximately 200 ml/100 g/min, and this limit is clearly approached in young anemic SCD patients, as shown by our data and several other methods. These patients, thus, are precariously poised at the edge of hemodynamic insufficiency. The most effective cerebral mechanism of increasing oxygen supply, and the one most often used as primary protective mechanism, is vasodilatation. As this mechanism is exhausted, any additional requirement for oxygen supply (e.g., increased metabolic demand, hypotensive episode, or local embolic event) must be satisfied by increased oxygen extraction, which is less efficient, less easily employed, and with a lower margin of safety than vasodilatation. The risk of stroke is, therefore, extremely high. Transfusion restores cerebrovascular reserve by reducing the excessive vasodilatation (Venketasubramanian et al, 1994), thus allowing the cerebral vasculature to meet future challenges.
These finding offer a different interpretation of observations from long use of TCD in SCD. Although initially elevated TCD velocity was thought to be due to vessel stenosis, we now suggest that TCD velocity is elevated primarily on the basis of flow disregulation. It is likely that the disruption of hemodynamics is the first event, followed coincidentally or secondarily by vessel stenosis. Strokes in SCD mainly reflect distal insufficiency due to exhaustion of vasomotor reserve.
Footnotes
Acknowledgements
This work was partially supported by NIH grant P60 HL28381 and the Maimonides Research Foundation. Some of these data were presented at the 14th International Symposium on Cerebral Blood Flow and Metabolism, Bologna, Italy, May 28 to June 1, 1989.
None.