
Abstract
Recently, a universal, simple, and fail-safe mechanism has been proposed by which cerebral blood flow (CBF) might be coupled to oxygen metabolism during neuronal activation without the need for any tissue-based mechanism. According to this concept, vasodilation occurs by local erythrocytic release of nitric oxide or ATP wherever and whenever hemoglobin is deoxygenated, directly matching oxygen demand and supply in every tissue. For neurovascular coupling in the brain, we present experimental evidence challenging this view by applying an experimental regime operating without deoxy-hemoglobin. Hyperbaric hyperoxygenation (HBO) allowed us to prevent hemoglobin deoxygenation, as the oxygen that was physically dissolved in the tissue was sufficient to support oxidative metabolism. Regional CBF and regional cerebral blood oxygenation were measured using a cranial window preparation in anesthetized rats. Hemodynamic and neuronal responses to electrical forepaw stimulation or cortical spreading depression (CSD) were analyzed under normobaric normoxia and during HBO up to 4 ATA (standard atmospheres absolute). Inconsistent with the proposed mechanism, during HBO, CBF responses to functional activation or CSD were unchanged. Our results show that activation-induced CBF regulation in the brain does not operate through the release of vasoactive mediators on hemoglobin deoxygenation or through a tissue-based oxygen-sensing mechanism.
Keywords
Introduction
Changes in neuronal activity are tightly coupled in both the spatial and temporal domains to changes in regional metabolism, regional cerebral blood flow (rCBF), and regional cerebral blood oxygenation (rCBO): ‘neurovascular coupling.’ Modern neuroimaging methods, such as functional magnetic resonance imaging (fMRI) and positron emission tomography, map brain activity with high spatial resolution by measuring changes in rCBF or rCBO. Although neurovascular coupling has been investigated for almost 120 years, the cellular and molecular mechanisms that couple neuronal activity to metabolism and blood supply are still incompletely understood. The ‘metabolic’ hypothesis implicates vasoactive products of metabolism, which are released from activated neurons and glial cells, such as H+, or adenosine. The ‘neuronal’ hypothesis, on the other hand, posits that neuronal energy demand is communicated to the vasculature (either directly or through astrocytes) in an anticipatory, feed-forward manner by vasoactive neurotransmitters or products of synaptic signaling, such as K+ or nitric oxide (NO) (Iadecola, 2004).
The additional oxygen consumed during functional brain activation is delivered by increased concentration of oxygenated hemoglobin (oxy-Hb), which becomes deoxygenated in this process. It is tempting to postulate that hemoglobin deoxygenation might be the master regulator of organ blood flow, as such a mechanism would potentially be fail-safe and independent of tissue factors.
Indeed, three major hemoglobin deoxygenation-based mechanisms have recently been postulated as universal regulators of tissue perfusion (for an overview, see Figure 1): Stamler et al (1997) have shown that in the lung the potent vasodilator NO binds to the β-93 cysteine moiety (βCys93) of oxy-Hb, forming S-nitroso-hemoglobin (SNO-Hb). Oxygenated blood then carries SNO-Hb to the tissue, where oxygen consumption induces allosteric transition of hemoglobin and release of NO on hemoglobin deoxygenation. Bound to thiols within the erythrocyte, NO is shuttled to the proximity of vascular smooth muscle cells, in which it induces guanylate-cyclase-dependent vasorelaxation. A second, recently described hemoglobin-deoxygenation-based mechanism to regulate organ perfusion postulates the release of NO from nitrite in an oxygen-consumption-dependent manner by the nitrite reductase activity of deoxygenated hemoglobin (deoxy-Hb) (Cosby et al, 2003). Finally, organ perfusion may be regulated by ATP, a potent vasodilator, released from erythrocytes depending on the amount of conversion of oxy-Hb to deoxy-Hb (Dietrich et al, 2000; Gonzalez-Alonso et al, 2002). A characteristic shared by all three mechanisms is that whenever and wherever oxygen is released from hemoglobin, a vasodilator is delivered (NO or ATP). This co-release of oxygen and vasodilator(s) might adapt blood flow to local hypoxia or local changes in oxygen metabolism in any tissue of any organism with hemoglobin-like oxygen carriers, independent of local tissue factors.
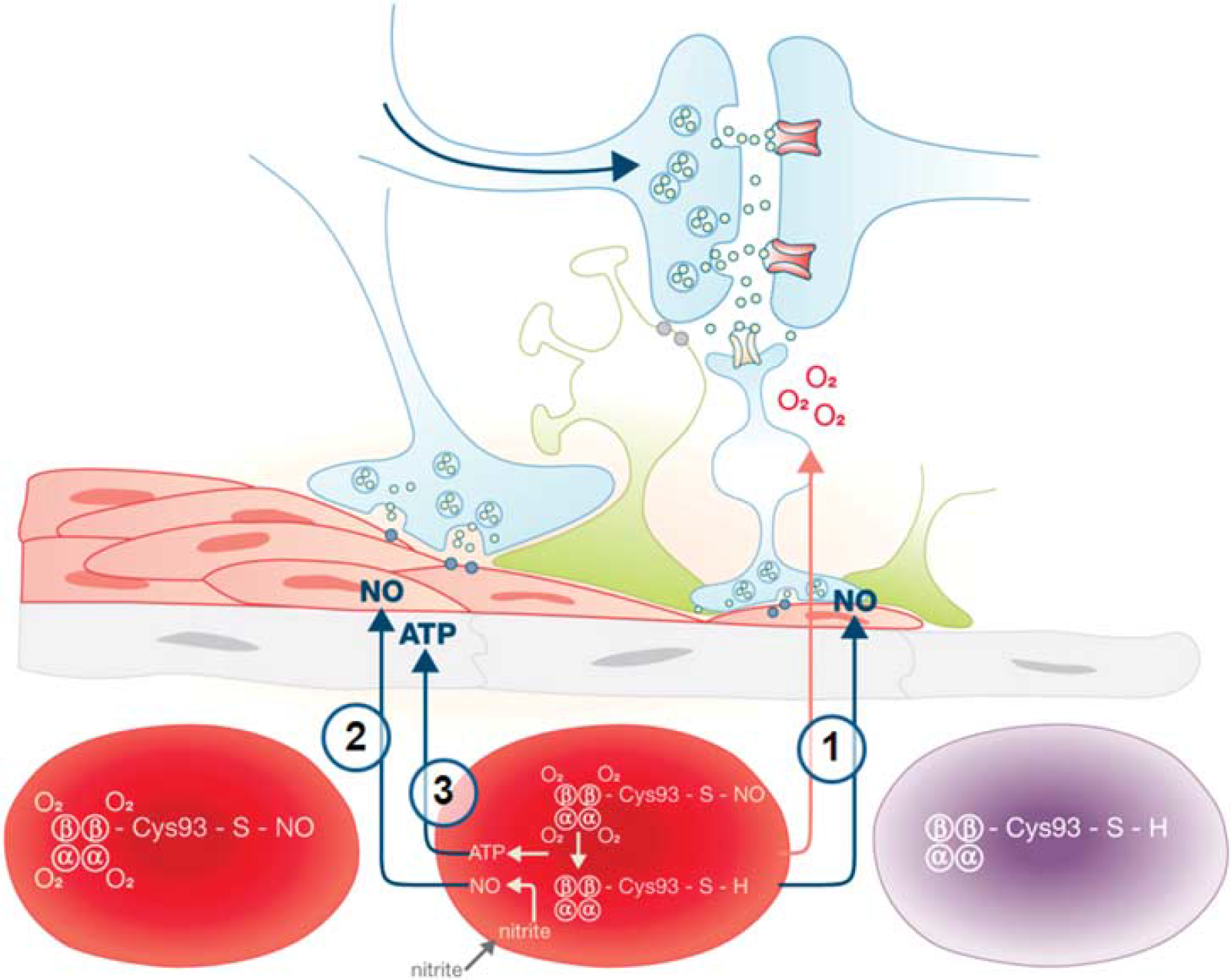
Schematic overview of hemoglobin deoxygenation-based mechanisms of blood flow regulation to increased oxygen demand: Recently, several elegant mechanisms of oxygen metabolism-induced delivery of the vasoactive molecules nitric oxide (NO) and ATP have been proposed for the coupling of tissue metabolism to blood flow in any tissue. Model 1: The potent vasodilator NO binds to the β-93 cysteine moiety (βCys93) of oxygenated hemoglobin (oxy-Hb), forming S-nitroso-hemoglobin (SNO-Hb). Oxygenized blood carries SNO-Hb to the tissues, where increased tissue oxygen consumption induces allosteric transition of hemoglobin and release of NO on hemoglobin deoxygenation (Stamler et al, 1997). Model 2: NO is released from nitrite in an oxygen-consumption-dependent way by the nitrite reductase activity of deoxygenated hemoglobin (deoxy-Hb; Cosby et al, 2003). Model 3: ATP is released by erythrocytes depending on the amount of conversion of oxy-Hb to deoxy-Hb (Dietrich et al, 2000; Gonzalez-Alonso et al, 2002).
Whether these elegant hemoglobin deoxygenation-based mechanisms have a role in regulation of blood flow in the brain has, surprisingly, never been tested experimentally during neuronal activation. In principle, SNO-Hb measurements could provide insights into whether hemoglobin delivers NO for tissue blood flow regulation. However, even in more easily accessible organs than the brain, this has led to controversial results (Gaston and Hare, 2003; Gladwin and Schechter, 2004; Schechter and Gladwin, 2003; Singel, 2003; Stamler, 2003), and detection of local arteriovenous gradients of the proposed mediators (SNO-Hb and ATP) is not feasible in the brain. In addition to methodological difficulties, the measurements of SNO-Hb, nitrite, or ATP would not allow researchers to establish a causal functional role for these molecules in blood flow regulation. Even recently presented data on new transgenic mouse models lacking the Hb-βCys93 residue were not able to solve the controversy (original publication Isbell et al, 2008; controversial discussion Patel and Townes, 2008; Stamler et al, 2008).
We have therefore opted to use hyperbaric hyperoxygenation (HBO) to test whether neuronal activation-induced vasodilation depends on the delivery of vasodilators during hemoglobin deoxygenation. During HBO with 100% oxygen concentration of gas at ≥ 3 standard atmospheres of absolute pressure (ATA; 1 ATA = 1.013 × 105 Pa), oxygen supply to tissue is provided entirely through oxygen physically dissolved in the blood: The periarteriolar O2 gradient is eliminated, no allosteric transition of oxy-Hb to deoxy-Hb occurs, and therefore SNO-Hb or nitrite remains preserved and no ATP is released by erythrocytes. When hemoglobin deoxygenation is prevented, the blood flow response should be blocked. In our experiments in anesthetized rats, we recorded CBF increases elicited by somatosensory stimulation during HBO. As functional activation leads to only small increases of oxygen metabolism, we also used the strong stimulus of neuronal metabolism and oxygen consumption, cortical spreading depression (CSD), and measured CSD-evoked CBF increases during HBO. We found that despite clamping brain oxy-Hb at 100%, somatosensory stimulation and CSD-induced CBF increases remained unchanged, clearly ruling out any significant contribution by hemoglobin deoxygenation- or tissue-oxygen-sensor-dependent mechanisms to neurovascular coupling.
Materials and methods
Animals
The experiments were approved by the ‘Landesamt für Arbeitsschutz, Gesundheitsschutz und technische Sicherheit Berlin’ (G 0133/02) and performed according to national and institutional regulations. A total of 35 male Wistar rats (250 to 380 g) were used for the following studies: functional activation study = 20 and CSD study = 15.
Animal Preparation
The animals were anesthetized with isoflurane (in 70% N2O, 30% O2) and artificially ventilated. Body temperature was continuously measured by a rectal probe and kept constant using a heating pad. The left femoral artery and vein were cannulated for arterial blood gas analysis, arterial blood pressure monitoring, and infusion of physiologic saline solution. The animals were placed in a stereotactic frame, the skull was removed, and a cranial window (dura mater intact) closed with a glass coverslip was implanted over the right somatosensory cortex. The space beneath the coverslip was filled with artificial cerebrospinal fluid (aCSF). Somatosensory evoked potentials (SEPs) (functional activation study) were measured through an epidurally placed Ag/AgCl ball electrode and averaged using a commercial EEG recording unit (Nihon Kohden Neuropack Monitor, Tokyo, Japan). In the CSD study, direct current (DC) potentials were measured through an epidurally placed Ag/AgCl ball electrode connected to an amplifier (Jens Meyer, Munich, Germany). A second open cranial window (small burr hole) was rostrally implanted over the parietal cortex in the CSD study to elicit CSD by KCl application. After surgery, anesthesia was switched to the α-chloralose–HBC complex (45 mg per kg body weight as intravenous (i.v.) bolus injection, followed by continuous i.v. infusion of 45 mg/kg per h as necessary). In the functional activation study, neuronal activation was induced by electrical sensory stimulation of the left forepaw (stimulation period 16 secs at 3 Hz; single stimulus duration 0.3 ms; intensity 1.2 to 1.6 mA; interstimulation interval 60 secs). In the CSD study, short-lasting trains of 3 to 5 CSDs were elicited at the frontal open window by a droplet of KCl (160 to 180 mmol/L), which was removed by repeated gentle flushing as soon as the first CSD wave was detected by DC potential shift and CBF response. HBO was induced in a hyperbaric chamber (Decompression Chamber Systems, Dräger Safety AG & Co. KGaA, Lübeck, Germany) flushed with 100% O2 and pressurized to 3 or 4 ATA, respectively. The hyperbaric chamber was especially constructed to afford space for the animal without additional space for the respirator or infusion pumps. Mechanical ventilation and i.v. infusion were therefore not feasible in the closed chamber during HBO periods. The animals breathed spontaneously within seconds after disconnection from the ventilator. Before interruption of continuous i.v. infusion, a bolus of α-chloralose–HBC complex (0.5 mL) was injected to maintain adequate anesthesia levels during HBO/sham HBO periods. Body temperature in the chamber was recorded and kept constant within the physiologic range by chamber heating.
Arterial blood gases were analyzed before each stimulation block or CSD block under normobaric normoxia at baseline and after HBO. In animals of the functional activation study, blood gases were also analyzed during HBO by a catheter originating from the femoral artery advanced through one of the pressure-resistant ports in the chamber wall. Systemic arterial blood pressure was recorded continuously with the exception of the period of hyperbaric pressure, during which recording was technically impossible.
After the experiments, the animals were killed in deep anesthesia by intravenous administration of concentrated KCl.
The experimental setup is illustrated in Figures 2A–2C.
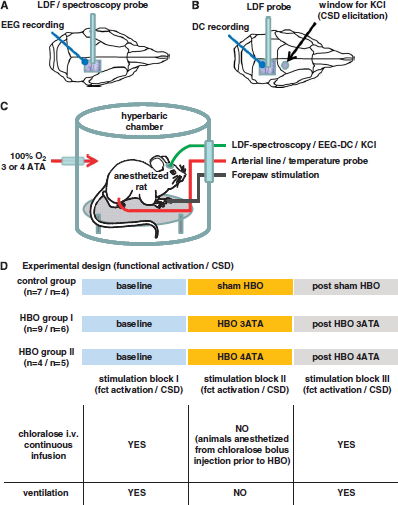
Experimental setup. (
While establishing the HBO chamber, in one animal, oxygenation of arterial blood of the systemic circulation was measured in the chamber at 3 ATA in the femoral artery catheter using an oxygen electrode (solid-state small needle electrode coupled to a LICOX—pO2 monitor, Integra NeuroSciences GmbH, Ratingen, Germany). In this animal, no CBF data were recorded.
Measurement of Cerebral Blood Flow and Blood Oxygenation
Changes in rCBF were measured through the cranial window by laser-Doppler flowmetry (LDF, Perimed AB, Järfälla, Sweden) and were recorded continuously with a time resolution of 100 ms. In four animals of the functional activation study (three of the 3 ATA group and one of the 4 ATA group), hemoglobin oxygenation changes in the somatosensory cortex were recorded in addition to CBF through the cranial window with an instrument and analysis which has been described in detail elsewhere (Royl et al, 2008). The device consisted of separate channels for measurement of CBF based on LDF and of hemoglobin oxygenation based on reflection spectroscopy. Of these four animals, the LDF data of two animals with 3 ATA HBO had to be discarded because of laser instability with significant and rapid baseline drifts. Therefore, the data on rCBF changes from seven out of nine animals in the 3 ATA group of the functional activation study were analyzed.
Experimental Design
The experimental design is illustrated in Figure 2D. Briefly, under normobaric normoxia (baseline condition, functional activation/CSD block I), rCBF responses to functional activation or CSD were recorded in each group. The animals were then transferred to the chamber; the chamber was flushed with 100% oxygen for 5 to 10 mins before closing, and was pressurized to 3 or 4 ATA, within 10 to 15 mins. Regional CBF and rCBO responses to functional activation or CSD were recorded again under hyperbaric hyperoxia (HBO condition, functional activation/CSD block II). The pressure within the chamber was then slowly brought back to baseline and rCBF responses to functional activation or CSD were recorded starting 20 mins after HBO (post-HBO condition, functional activation/CSD block III). In the control group, the animals were treated accordingly, with the exception that the chamber was flushed with room air and not pressurized (21% O2 at 1 ATA, sham HBO).
Data Analysis
Cerebral blood oxygenation:
In four animals of the functional activation study, averaged responses in oxy-Hb, deoxy-Hb, and total hemoglobin (total-Hb; sum of oxy-Hb and deoxy-Hb, reflecting blood volume) at normoxia, during and after HBO, were calculated and presented as concentration change from resting values directly before stimulation. Owing to the low number of animals, no statistical analysis of hemoglobin oxygenation changes was carried out in this data set.
Cerebral blood flow:
The rCBF responses to functional activation or CSD under hyperbaric hyperoxia (HBO) and after hyperbaric hyperoxia (post-HBO) were compared with responses elicited in each animal during baseline conditions under normobaric normoxia (baseline) in the same group. In the functional activation study, CBF data are presented as the average of 8 to 12 stimulus trains during baseline, HBO and post-HBO, respectively. Regional CBF changes in the functional activation study are calculated as percentage increase from resting flow before stimulation onset. For quantification, functional activation-induced rCBF increases were calculated as the mean percentage increase of the period of 5 to 16 secs after stimulation onset (plateau phase of the CBF response). At the higher CBF values that occur during CSD, LDF-CBF overestimates absolute blood flow and is therefore no longer linearly correlated with CBF measured by the gold standard of autoradiography (Dirnagl et al, 1989). Therefore, rCBF changes during CSD in arbitrary units (AU) of LDF were used for further analysis. CSD-induced rCBF increases are presented as the maximum amplitude of the CBF response during CSD.
Neuronal activity:
Neuronal activity was assessed by SEP and DC potential recording. For comparison of functional changes of neuronal activity during baseline conditions and during as well as post-HBO periods, the absolute amplitude of the averaged SEP (P1 to N1) or the absolute value of the maximal negative shift of the DC potential during spreading depression was used for further analysis.
Data normalization:
For quantitative analysis, resting CBF as well as the mean responses of CBF and neuronal activity (SEP and DC potential amplitude) during and after HBO were normalized to the values under baseline conditions in each animal, to correct for differences in resting CBF and for differences in stimulation responses in the neuronal and vascular parameters during baseline conditions between animals.
Statistical Analysis
For averaged time courses of LDF, oxy-Hb, deoxy-Hb, and total-Hb, mean values were calculated from all animals tested. Quantified data are presented as vertical boxes with error bars (median, 10th, 25th, 75th, and 90th percentiles, single values included). Comparisons between baseline, HBO, and post-HBO are conducted using Kruskal–Wallis one-way analysis of variance on ranks. When differences were detected, we used Dunn's method for multiple comparisons versus baseline as post hoc test (SigmaStat 2.0, SPSS Inc, Chicago, IL, USA; statistical software). P < 0.05 was considered as statistically significant.
Results
Systemic Physiologic Parameters Remained Within the Physiologic Range
With the exception of pO2 during HBO, all systemic parameters were within physiologic ranges during the experiments. A slight decrease in arterial pO2 was detected in the control group during and after sham HBO (animals transferred to hyperbaric chamber, experiment conducted at normobaric normoxia) as was a slight increase in arterial pCO2 in the HBO groups during the period of HBO (see Table 1).
Physiological parameters
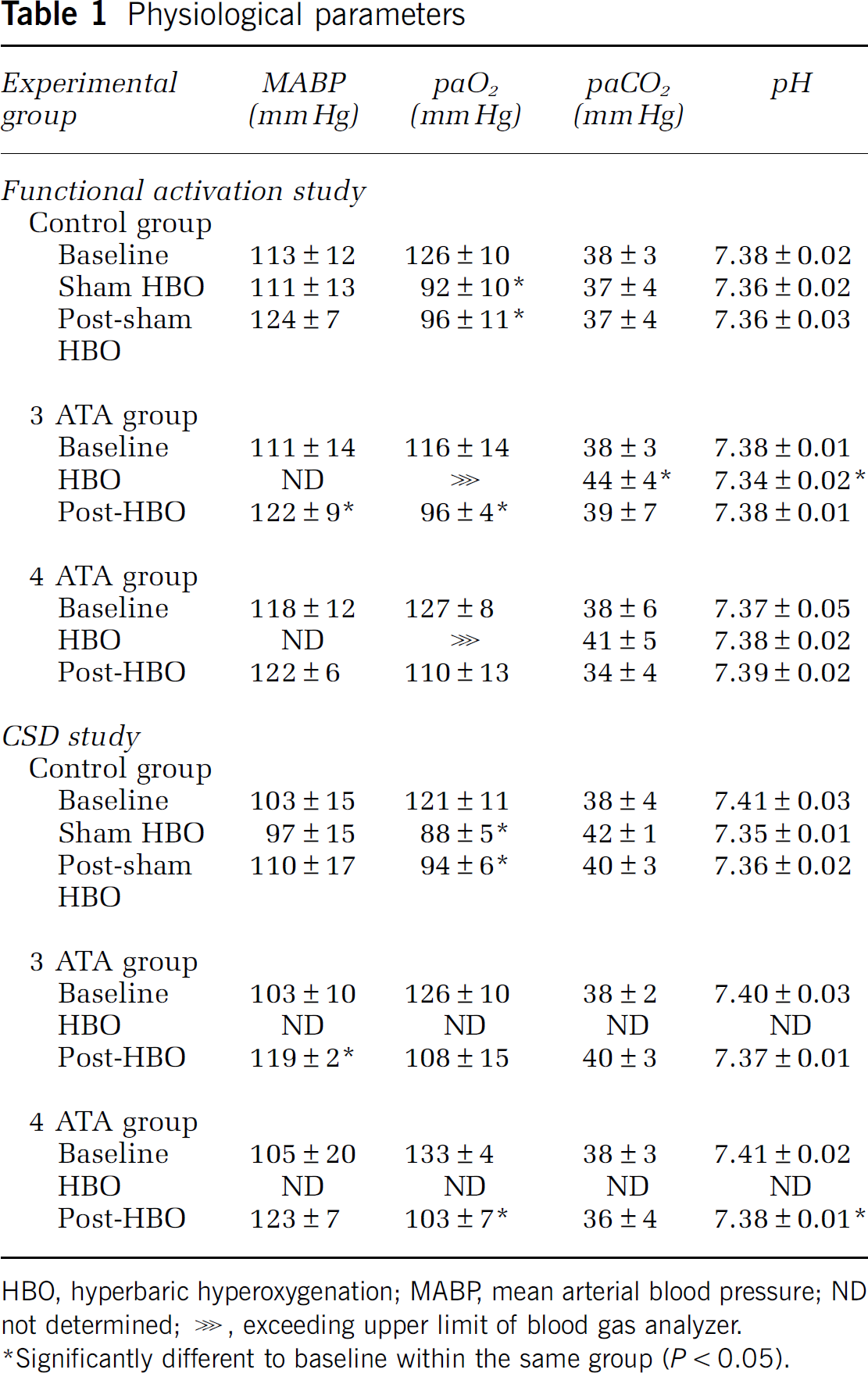
HBO, hyperbaric hyperoxygenation; MABP, mean arterial blood pressure; ND not determined; ⋙, exceeding upper limit of blood gas analyzer.
Significantly different to baseline within the same group (P < 0.05).
In one animal, systemic arterial pO2 measured with an oxygen electrode within the chamber at 100% oxygen and 3 ATA exceeded the upper detection limit of the electrode of 2000 mm Hg. This confirms the effectivity of flushing the chamber with 100% oxygen at a pressure of 3 ATA. The value calculated for systemic arterial pO2 under these conditions should be ∼2000 mm Hg, assuming an atmospheric pressure of 760 mm Hg under normal conditions at 1 ATA and considering the arterial pCO2 and the vapor pressure of water.
Resting Cerebral Blood Flow Slightly Increased During and After Sham Hyperbaric Hyperoxygenation or Hyperbaric Hyperoxygenation
In the functional activation study, a slight increase in resting CBF was detected in the control group (Supplementary Figure S1a, Supplementary Material) as well as in the HBO groups (Figure 3A). In the CSD study, resting CBF showed the same tendency toward higher values during sham HBO (Supplementary Figure S1a, Supplementary Material) or HBO (Figure 3B). The slight increase in resting CBF was most likely attributable to a slight increase in paCO2. We could not counteract the paCO2 increase through adjustment of ventilation, because our pressure chamber does not allow for mechanical ventilation of the animals.
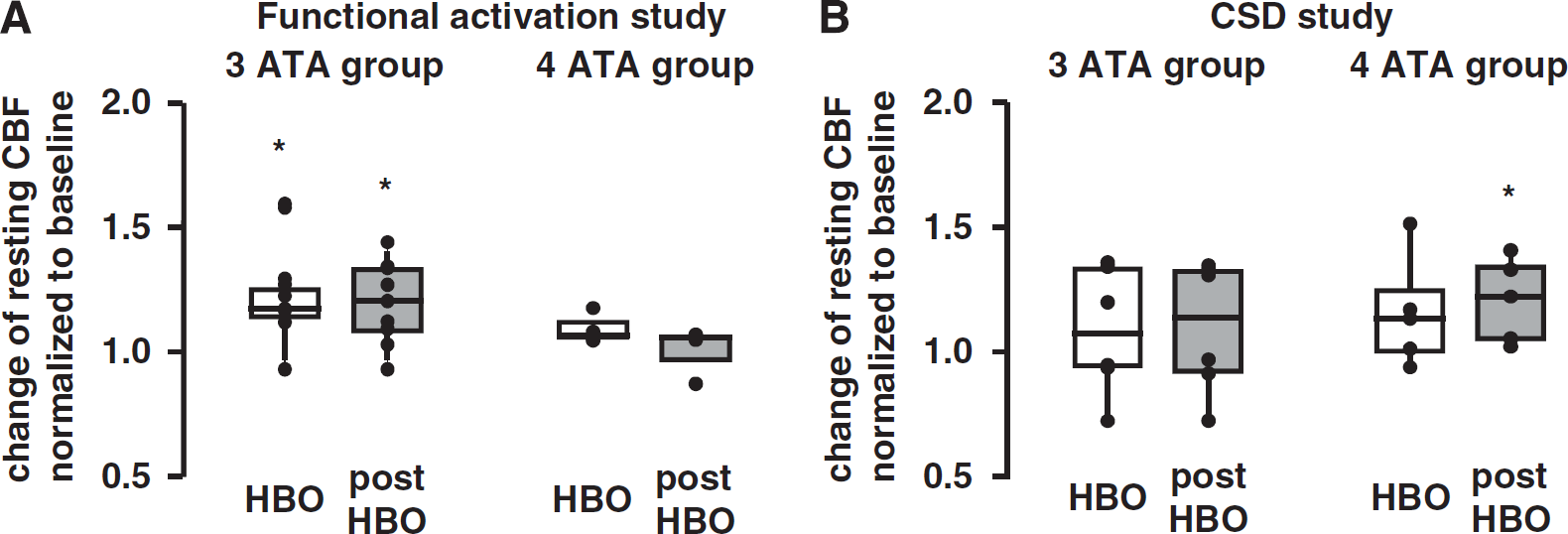
Effect of hyperbaric hyperoxygenation (HBO) on resting cerebral blood flow (CBF). In the (
Stimulation-Induced Neuronal and Cerebral Blood Flow Responses Remained Stable in the Control Groups
Evoked neuronal activity and stimulation-induced CBF responses during functional activation and CSD remained unaltered in the control groups of both studies during sham HBO and post-sham HBO (see Supplementary Figure S1b and c, Supplementary Material).
Deoxygenated Hemoglobin Response to Functional Activation Disappeared During Hyperbaric Hyperoxia
In four animals of the functional activation study, we applied optical spectroscopy in addition to LDF to measure hemoglobin oxygenation changes. In all animals tested, the deoxy-Hb changes disappeared during HBO at 3 or 4 ATA (see Supplementary Figure S2, Supplementary Material).
Hyperbaric Hyperoxia Showed no Effect on Neurovascular Coupling During Functional Activation or Cortical Spreading Depression
Stimulated neuronal activity (SEP: typical example in Figure 4A, quantified results in Figure 4B; DC potentials: typical example in Figure 5A, quantification in Figure 5B) did not change during HBO at 3 or 4 ATA compared with their respective baseline. SEP or DC amplitudes and temporal dependence remained constant, showing cellular activation to functional stimulation or CSD comparable under baseline condition and HBO. Even during 4 ATA, no seizure activity was observed in the anesthetized animals.
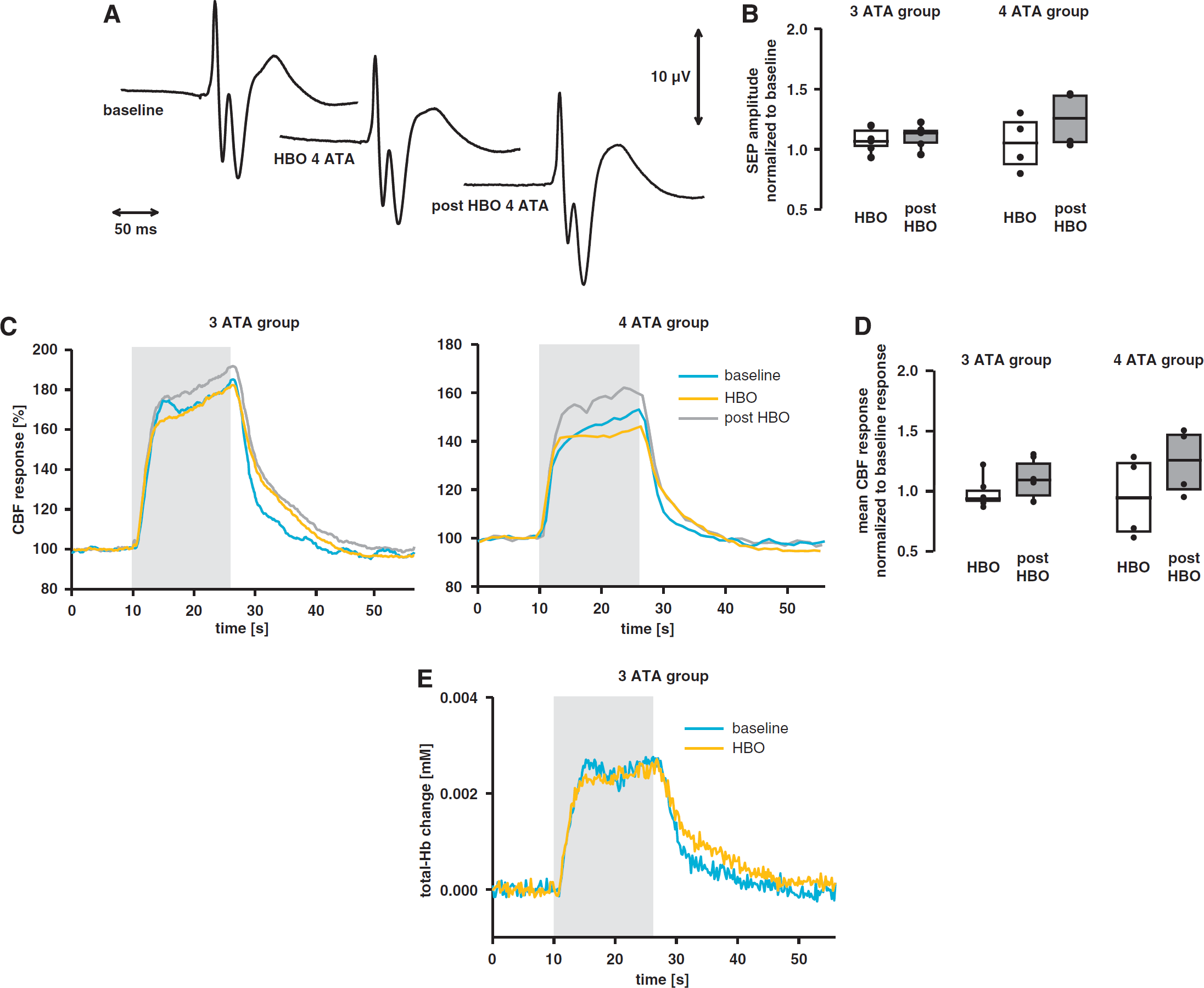
No effect of hyperbaric hyperoxygenation (HBO) on neuronal activity and neurovascular coupling during functional activation. (
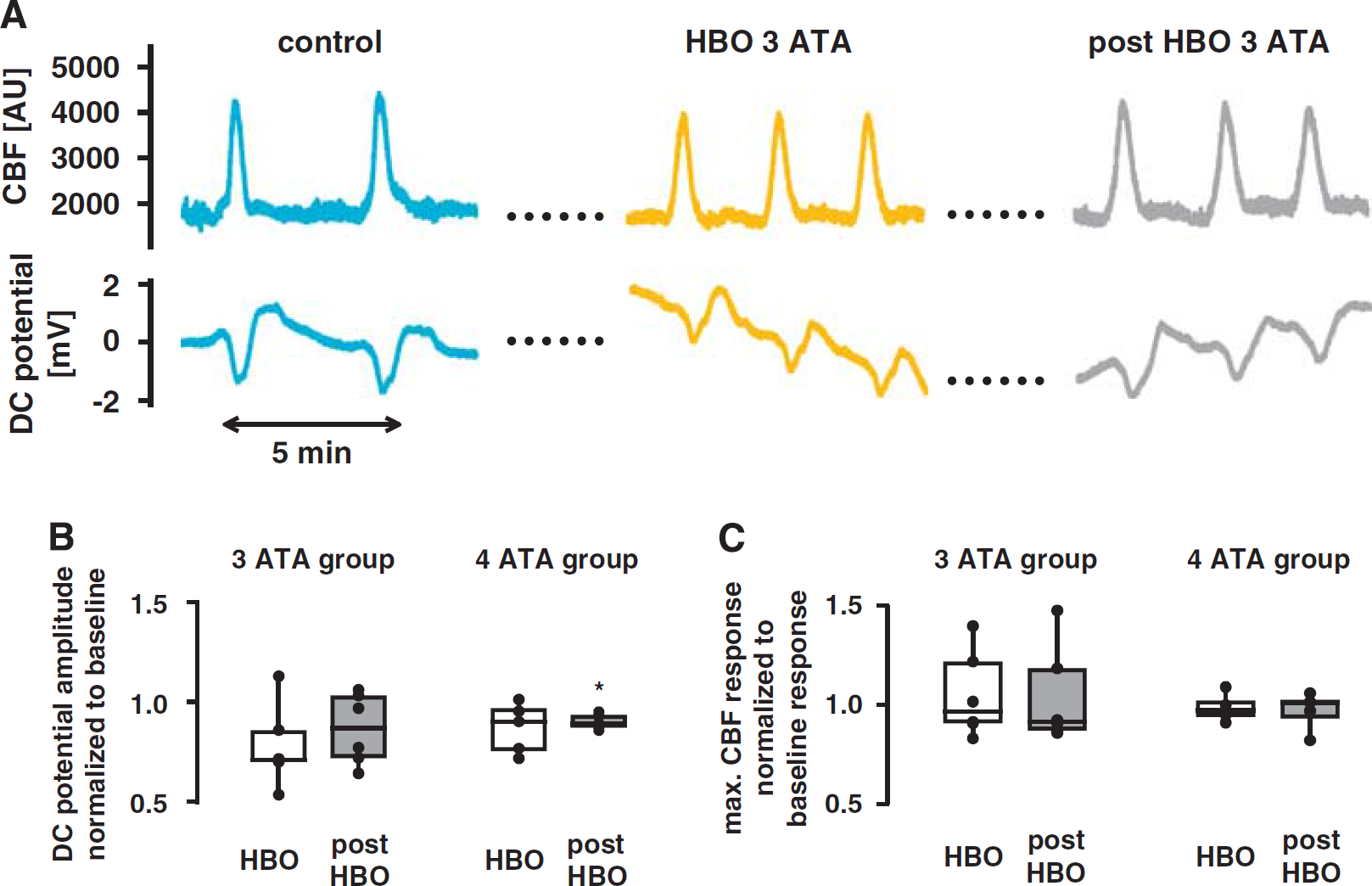
No effect of hyperbaric hyperoxygenation (HBO) on neuronal activity and neurovascular coupling during cortical spreading depression. (
Excessive oxygen supply during HBO up to 4 ATA had no effect on the CBF responses to functional activation or CSD. Neither the dynamic course (Figure 4C) nor the mean amplitude (Figure 4D) of the blood flow increase during functional activation showed any difference between baseline condition, HBO, and post-HBO in any of the groups tested. In addition, corpuscular blood volume changes to functional activation remained constant during HBO, as shown by optical spectroscopy at 3 ATA (Figure 4E). Even during the more pronounced metabolic stimulus of CSD, neither the dynamic time courses (typical example Figure 5A) nor the maximal increases of CBF (quantified results Figure 5C) changed during or after HBO compared with their baseline conditions.
Discussion
In this study, we tested whether blood-borne mediators released from hemoglobin on deoxygenation might, at least partially, explain blood flow increases during increased neuronal activity and increased oxygen metabolism in the brain. To this end, we functionally activated the primary somatosensory cortex (SI) in rats by electrical sensory stimulation of the contralateral forepaw, or by elicitation of CSD, in a hyperbaric chamber filled with 100% O2 at 3 or 4 ATA. Under these conditions, oxygen supply to brain tissue is provided entirely by physically dissolved oxygen, and no deoxygenation of hemoglobin occurs. Using LDF and optical spectroscopy, rCBF and rCBO were recorded through a closed cranial window overlying the SI cortex during normobaric normoxia, HBO and after return to normobaric normoxia. Neuronal activity was monitored by recording of SEPs and DC potentials using epidural electrodes. Contrary to the hypothesis, HBO did not change the rCBF or corpuscular blood volume responses to functional activation or the rCBF responses to CSD (Figures 4 and 5), and rCBF remained tightly coupled to neuronal activity during excessive oxygen availability.
During Hyperbaric Hyperoxia, Physically Dissolved Oxygen Fully Supports Increases in Stimulation-Induced Oxygen Metabolism
Under physiologic conditions, depending on hematocrit, per 100 mL blood around 20 mL O2 are bound to hemoglobin and only 0.3 mL O2 are physically dissolved. Under resting conditions, an overall amount of 5 to 6 mL O2/100 mL of blood is consumed in rat brain, as estimated by Vovenko (1999). These values largely depend on blood hemoglobin concentration, CBF, and CMRO2 (cerebral metabolic rate of oxygen), and might vary considerably between different experimental setups. During hyperbaric hyperoxia at 3 ATA, systemic arterial pO2 reaches ∼2000 mm Hg. At partial pressures > 3 ATA, brain concentrations of oxygenated hemoglobin and thus microcirculatory hemoglobin saturation reach a maximum (Meirovithz et al, 2007), the amount of physically dissolved oxygen significantly increases up to 7 mL/100 mL blood, and tissue pO2 in the brain increases to more than 400 mm Hg (Demchenko et al, 2005; Jamieson and Van den Brenk, 1962). As shown by Demchenko et al (2005), tissue pO2 values of above 400 mm Hg were reached already at 3 ATA when hyperoxygenation-induced decrease of resting CBF was prevented by acetazolamide application, which slightly elevated the arterial pCO2 up to 44 mm Hg. In this study, owing to spontaneous breathing in the hyperbaric chamber, arterial pCO2 and resting CBF were increased to values comparable with those achieved by Demchenko et al (2005), suggesting an equally elevated tissue pO2 of > 400 mm Hg. This increase in tissue O2 leads to a complete oxidation of cytochrome oxidase type aa3 (Hempel et al, 1977) and of mitochondrial NADH (Meirovithz et al, 2007). During HBO, oxygen consumption during neuronal activation could therefore be matched (3 ATA) or even exceeded (4 ATA) by the amount of physically dissolved oxygen. To verify that hemoglobin deoxygenation in brain microcirculation was indeed prevented by hyperbaric hyperoxia in our setup, rCBO was recorded by optical spectroscopy in some animals. Deoxy-Hb remained unchanged in response to functional activation during HBO in all animals tested at 3 or 4 ATA HBO, showing that hemoglobin deoxygenation can be prevented by HBO. When hemoglobin remains fully oxygenated, that is, clamped at 100%, no vasodilator release from hemoglobin, if existent at all, can occur.
Functional Activation-Induced Neurovascular Coupling in the Brain Operates Independent of Hemoglobin Deoxygenation or Tissue-Based Oxygen Sensors
Vasodilation to local tissue hypoxia may not be a direct effect of oxygen at the vascular wall. It has been shown that isolated cerebral arterioles only dilate to hypoxia when they are perfused with red blood cells (Dietrich et al, 2000). These findings suggest that erythrocytes release or bind vasodilators depending on the availability of perivascular oxygen, resulting in vasodilation or vasoconstriction. Indeed, several mechanisms of oxygen-dependent release of vasoactive substances have been proposed in the last decade, which might be responsible for the tight vascular coupling to oxygen metabolism and hence to activity and energy demand of the tissue. The common feature of these mechanisms is deoxygenation of hemoglobin during increased oxygen demand. As mentioned above, oxygen delivery to tissues occurs mainly through oxygen released from hemoglobin, as the amount of physically dissolved oxygen in blood is very low under normoxic conditions. During hemoglobin deoxygenation, either NO or ATP may be released, both of which are strong vasodilators (Cosby et al, 2003; Dietrich et al, 2000; Gonzalez-Alonso et al, 2002; Jia et al, 1996; Stamler et al, 1997). To functionally study hemoglobin deoxygenation-related mechanisms independent of specific mediators, the ideal approach is to investigate neurovascular coupling under conditions without conversion of oxy-Hb to deoxy-Hb. Our results showing unchanged CBF responses within the somatosensory cortex to sensory forepaw stimulation in anesthetized rats under HBO strongly argue against a functional role of blood-borne vasodilators in neurovascular coupling. Before our findings that excessive oxygen supply has no effect on neurovascular coupling, it had been shown in human subjects that the CBF response to physiologic activation was not altered by moderate hypoxia (Mintun et al, 2001). Taken together, these results suggest that the mechanism of CBF increase during physiologic brain activation is not dependent on a shortage of oxygen. Our findings also argue against a tissue oxygen-sensing mechanism, potentially at the level of mitochondria, matching blood flow to metabolic demands of oxygen, as recently proposed (Gjedde, 2002).
We have used a comparably strong forepaw stimulation that produced large increases in flow (60% to 80% increase in the 3 ATA HBO group), because in the short HBO period, extensive averaging of stimuli was not possible and large CBF responses can therefore reduce noise and increase the statistical power of our analysis. Therefore, one may wonder whether, during these strong hyperemic stimuli, different vasodilator mechanisms that overwhelm the purported hemoglobin-based mechanism are engaged. Owing to interindividual variations of CBF responses, a number of animals had smaller CBF responses. We have therefore reanalyzed the data of four animals (HBO at 3 or 4 ATA) with an average CBF response of 47% (Supplementary Figure S3, Supplementary Material), which is in good agreement with recent studies reporting blood flow responses in awake humans of 63% during visual or of 46% during motor stimulation (Donahue et al, 2009) or in anesthetized rats of 68% (fMRI; Stefanovic et al, 2007) or 36% (laser speckle contrast imaging; Sakadzic et al, 2009) during electrical forepaw stimulation. In accordance with the results obtained from all animals, the CBF responses during HBO in this subgroup of animals did not change compared with their respective baseline responses. This retrospective analysis showed no evidence that our results are confounded by the magnitude of the CBF responses. In addition, we have already shown in a previous pilot study that HBO does not affect neurovascular coupling to forepaw stimulations with 20% to 30% increases in CBF (Leithner et al, 2005).
It may be considered a limitation of our findings that we cannot completely exclude a role for Hb-S- or nitrite-derived NO or ATP under normoxic conditions that might be quantitatively compensated for by another pathway during HBO. However, it seems unlikely that during HBO one of the other known pathways or another still unknown pathway (Leithner et al, 2009) is more activated if hemoglobin deoxygenation-dependent vasodilators are blocked, leading to identical magnitude and time course of the CBF response.
During HBO, a slight increase of resting CBF was detected that was most likely attributable to a slight increase in paCO2. In principle, the CBF response may be determined by the baseline resistance/blood flow. To rule out that the increased resting CBF altered the CBF response to functional activation, we have reanalyzed the data of five animals (HBO at 3 or 4 ATA) with small paCO2 changes and thus stable resting CBF during HBO. As shown in Supplementary Figure S4, the CBF responses during HBO in this subgroup of animals did not change compared with their respective baseline responses, which is in accordance with the results obtained from all animals.
In summary, our data strongly argue against a neurovascular coupling mechanism depending on oxygen availability in the brain or blood. It is noted that this does not rule out matching oxygen delivery with tissue metabolism as an evolutionarily evolved function of neurovascular coupling, which may have developed as a feed-forward mechanism.
Hemoglobin Deoxygenation Does Not Regulate the Blood Flow Response to High Metabolic Demand of Oxygen (CSD)
The relatively small increase in oxygen metabolism within the somatosensory cortex of rats during functional activation (10% to 15%; Royl et al, 2008) may not be sufficient to release relevant amounts of the hemoglobin deoxygenation-dependent vasodilators. However, hemoglobin deoxygenation-based mechanisms may have a role in preventing tissue from moderate or severe hypoxia during intense metabolic stress. It was shown recently that the blood flow response in the heart and the diameter response of isolated thoracic aorta from the rabbit or mouse during severe hypoxia depend on O2-dependent delivery and release of NO from hemoglobin (Datta et al, 2004; Diesen et al, 2008; James et al, 2004).
We therefore tested whether hemoglobin deoxygenation-mediated coupling of blood flow and oxygen metabolism might be operative during the much stronger metabolic stimulus of CSD in the cerebral cortex. The significant increase in energy need during CSD is reflected in a 200% to 300% increase in glucose metabolism (Gjedde et al, 1981; Nedergaard and Astrup, 1986) and a 50% increase in oxygen metabolism (Mayevsky and Weiss, 1991), suggesting that the high ATP demand during CSD is largely met by oxidative metabolism. Very recently, it was shown that despite a marked blood flow increase, CSD was associated with local depletion of tissue O2 and marked neuronal swelling (Takano et al, 2007). In comparison with physiologic functional activation, CSD may therefore be even better suited to test for a hemoglobin deoxygenation-based mechanism of blood flow regulation in the brain.
However, CBF responses to the intense metabolic stimulus of CSD did not change during HBO.
Evidence Against Astrocyte-Dependent Mechanisms Inducing Vasoconstriction Under Hyperoxia
Within the last decade, a role for astrocytes in neurovascular coupling has been established. Astrocytic mediators released by glutamate uptake during neuronal activation may be involved in triggering the CBF response to functional activation (Filosa et al, 2004; Metea and Newman, 2006; Mulligan and MacVicar, 2004; Peng et al, 2002; Schummers et al, 2008; Zonta et al, 2003). Using a rat brain slice preparation, Gordon et al (2008) very recently showed a strong dependency of the astrocytic control of arteriolar diameters on brain tissue metabolism and oxygen supply. Their findings show that under conditions of low oxygen concentration (aCSF bubbled with 20% O2), astrocytes couple arteriolar tone to neuronal activation by prostaglandin E2-mediated and lactate- and adenosine-dependent vasodilation. In contrast, during conditions of high oxygen (aCSF bubbled with 95% O2), the same stimulus induced vasoconstriction. Applying these data to neurovascular coupling in vivo, a reduction in the CBF increase or even its conversion to a CBF decrease during tissue hyperoxygenation must be postulated. However, neither normobaric hyperoxia (Wolf et al, 1997) nor hyperbaric hyperoxia at 3 or 4 ATA (this study) was able to even reduce the normal vasodilation during functional activation in rats. It has to be kept in mind that studying blood flow regulation without blood flow (brain slice) excludes numerous potentially relevant effectors such as corpuscular and blood plasma flux- or flow-induced vascular tone.
Conclusion
In this study, we have shown that clamping brain hemoglobin oxygenation at 100% by HBO up to 4 ATA does not change the CBF response to physiologic functional activation or transient tissue depolarization during CSD. Our results indicate that the regional coupling of blood flow in the brain to physiologic or pathophysiologic neuronal activation is independent of the level of oxygen transported to the tissue, and that shortage of oxygen is not a driving force for vasodilation during increased neuronal activity. Matching blood flow to neuronal activity in the rat brain must involve mechanisms other than hemoglobin deoxygenation-dependent delivery of vasodilators. Several mechanisms have been taken into consideration within the last decade such as neuronal or astrocytic products of synaptic signaling (K+ or NO; Iadecola, 2004), activation of GABA interneurons (Kocharyan et al, 2007), or arachidonic acid metabolites from neurons (production of prostaglandin E2 by cyclooxygenase activation; Niwa et al, 2000) or astrocytes (production of epoxyeicosatrienoic acids by CYP450 epoxygenase; Peng et al, 2002). Together with previous findings on the lack of effect of hypoglycemia or hyperglycemia on neurovascular coupling (Powers et al, 1996; Wolf et al, 1997), our data suggest that rCBF responses to functional activation are regulated in a feed-forward manner, and not by a shortage of energy substrates. Our results therefore strongly support the ‘neuronal’ hypothesis of a neuronal or glial signal inducing vasodilation in an anticipatory manner.