
Abstract
The Rho signaling has an essential function in human immunodeficiency virus (HIV)-1-mediated disruption of the integrity of the blood–brain barrier (BBB). However, it is unknown how membrane domains, such as lipid rafts, can influence HIV-1-mediated activation of the Rho pathway and how these processes can affect the expression of the efflux transporters at the BBB level. This study is focused on the function of HIV-1 protein Tat in activation of the Rho signaling and upregulation of P-glycoprotein (P-gp) in human brain endothelial cells. Treatment with Tat markedly elevated GTP-RhoA levels and the potential downstream effectors, such as myosin phosphatase target subunit 1 and myosin light chain. In addition, Tat upregulated expression and promoter activity of P-gp as well as its efflux function. Inhibition of the Rho signaling cascade effectively blocked P-gp overexpression at the level of promoter activity. Disruption of lipid rafts by depletion of membrane cholesterol by methyl-beta-cyclodextrin, but not caveolin-1 silencing, also abolished Tat-mediated RhoA activation and P-gp upregulation. The present data indicate the critical function of intact lipid rafts and the Rho signaling in HIV-1-mediated upregulation of P-gp and potential development of drug resistance in brain endothelial cells.
Introduction
Membrane receptors and components of their signaling pathways are spatially organized in membrane microdomains called lipid rafts. Caveolae are a specific subset of lipid rafts that is characterized by the presence of caveolin proteins on the inner leaflet of the membrane bilayer. Endothelial cells express caveolin-1 and -2 proteins; however, caveolin-1 is the main isoform and knocking it down eliminates caveolae. Evidence indicates that lipid rafts and caveolae regulate numerous cellular processes, including cell polarity, protein trafficking, and signal transduction (Cohen et al, 2004). Lipid rafts contain specific sets of receptors and signaling proteins, such as G-protein-coupled receptors. Their activation results in the stimulation of heterotrimeric G-proteins and small guanosine 5′-triphosphate (GTPases), including the Rho proteins. The significance of the Rho signaling in maintaining the integrity of the blood–brain barrier (BBB) has recently been demonstrated. The Rho proteins act as a molecular switch, cycling between the active GTP-bound state and inactive guanosine 5′-diphosphate (GDP)-bound state (Sahai and Marshall, 2002). GTP-Rho proteins activate the Rho kinase and a number of downstream effector proteins, including the RhoA/Rho kinase (ROCK) (Rolfe et al, 2005), myosin-binding subunit of myosin light chain (MLC) and MLC phosphatase (MLCP). Of these proteins, MLCP appears to be a physiologic substrate for ROCK. For example, ROCK has been shown to phosphorylate and thus inhibit MLCP, causing the sustained smooth muscle contraction even after a decrease in Ca2+ concentrations. Rho proteins are involved in a range of cellular processes, such as regulation of the actin cytoskeleton, cell mobility, polarity, and proliferation (Coleman et al, 2004; Sahai and Marshall, 2002). In addition, RhoA and Rac1 can regulate occludin phosphorylation, stress fiber formation, and endothelial permeability (Wojciak-Stothard et al, 2005). However, the function of the Rho signaling in regulation of efflux transporters, such as P-glycoprotein (P-gp) is unknown.
P-glycoprotein is the 170-kDa transmembrane glycoprotein encoded by the human multidrug resistance MDR 1 gene, and a member of the ATP-binding cassette transporter superfamily. P-glycoprotein functions as an ATP-dependent exporter of xenobiotics from cells. It constitutes the main efflux transport system in cells and tissues with excretory functions such as brain capillary endothelial cells, intestine, liver, kidneys, placenta, testis, and peripheral blood cells. It is believed that P-gp has evolved as a biological defense mechanism against the entry of toxic substances from the gut into the blood and for the protection of the brain. The location of P-gp in brain capillaries is essential for the proper function of the BBB as several drugs exhibit higher accumulation in the brains of P-gp knockout mice [reviewed by Miller et al (2008)]. Importantly, evidence indicates that P-gp can be upregulated in human immunodeficiency virus (HIV)-infected patients (Langford et al, 2004).
Highly active antiretroviral therapy (HAART) has helped transform HIV infection from a fatal illness into a chronic and manageable condition. However, only approximately half of the patients achieve full success with HAART owing to drug resistance, difficulties in adhering to complex medication regiments, or intolerable side effects. Despite its success in decreasing viral loads in blood, HAART has not eliminated HIV infection of the central nervous system (CNS). Indeed, various forms of neurocognitive impairment occur in 50% or more of HIV-1-infected individuals (Cysique et al, 2004).
The HIV-1 invades the brain early in the course of systemic infection. However, most antiretroviral drugs do not penetrate the brain effectively; thereby, HIV can be latently preserved in the CNS, generating an important viral reservoir [reviewed by Ellis et al (2007)]. Several mechanisms are responsible for the restricted movement of many antiretroviral drugs from the circulation into the CNS. For example, antiretroviral drugs, especially protease inhibitors (e.g., indinavir, nelfinavir, and saquinavir), are substrates for P-gp expressed in brain endothelial cells. The active removal of antiretroviral drugs from the CNS results in viral replication in the brain, facilitates viral mutations, and thus, precipitates failure of antiretroviral therapy (Washington et al, 2000).
Studies from our and other laboratories indicate that several mechanisms of HIV-1-induced vascular pathology can be reproduced by treatment with the HIV-1 protein, Tat. This protein is produced and released by HIV-1-infected cells and circulating Tat is detected in HIV-1-infected patients (Xiao et al, 2000). Exposure of brain endothelial cells to Tat results in the disruption of tight junction proteins through activation of the Ras/mitogen-activated protein kinase pathway (Zhong et al, 2008). Tat also increases cell proliferation, angiogenesis, and the development of Kaposi sarcoma via its direct effects on Kaposi sarcoma cells (Toschi et al, 2006). Furthermore, Tat can induce the expression of inflammatory mediators such as tumor necrosis factors-α and monocyte chemoattractant protein-1 in mouse brains (Toborek et al, 2003).
Because of the importance of P-gp as an efflux transporter that limits entry into the brain of antiretroviral drugs, the aim of this study is to determine signaling mechanisms involved in this process. Our studies indicate that Tat can increase P-gp expression by activation of the Rho/ROCK signaling pathway. It appears that intact lipid rafts, but not caveolae, provide the essential signaling platform for these effects. This study is the first report on the function of lipid rafts and the Rho pathway in regulation of P-gp expression.
Materials and methods
Materials
Fetal bovine serum (FBS) was purchased from HyClone (Logan, UT, USA). Endothelial Cell Basal Medium-2 (EBM-2 medium) and EGM-2 SingleQuots were from Lonza (Walkersville, MD, USA). Polyclonal antibodies directed against phospho-MLC 2 (Ser19) and MLC 2 were from Cell Signaling Technologies (Beverly, MA, USA). Anti-MYPT-1 antibody was from Affinity BioReagents (Golded, CO, USA). Anti-phospho-MYPT-1 (Thr853), anti-Rho A, anti-mouse-IgG-HRP, anti-rabbit-IgG-HRP, anti-goat-IgG-HRP antibodies were from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Anti-P-gp monoclonal antibody (C219) was from EMD Biosciences (Gibbstown, NJ, USA). The ROCK inhibitor Y27632, methyl-beta-cyclodextrin (MβCD) and anti-actin antibody were purchased from Sigma (St. Louis, MO, USA). ML-9 was from Cayman Chemical (Ann Arbor, MI, USA), and RhoA activation assay kit was obtained from Millipore (Temecula, CA, USA).
The HIV-1 Tat protein was purified as described earlier (Ma and Nath, 1997). To determine specificity of Tat-induced effects, Tat was immunoabsorbed using anti-Tat antibody conjugated to protein-A/G plus-agarose (Santa Cruz Biotechnology) and centrifuged to remove precipitated proteins. This antibody–absorption mixture was called AA-Tat and was used as negative control in this study.
Cell Cultures and Tat Treatment
Human cortical microvessel endothelial (hCMEC/D3) cells were kindly provided by Dr Pierre-Olivier Couraud (Institut COCHIN, Paris, France). The cells were cultured in EBM-2 medium supplemented with EGM-2 SingleQuots and 5% FBS (EBM-2 completed medium) as described earlier (Zhong et al, 2008). Before treatments, the cells were maintained in EBM-2 medium without FBS and EGM-2 SingleQuots for 24 h.
Concentrations of Tat in HIV-infected patients can reach the range of nanograms per ml of serum (Xiao et al, 2000). Therefore, cells were treated with Tat at the levels between 1 and 100 nmol/L in this study. Such concentrations of Tat did not alter viability of hCMEC/D3 cells as assessed by MTT assay (data not shown) and are consistent with literature data (Rumbaugh et al, 2006). Tat at the level of 100 nmol/L markedly decreased tight junction protein expression in hCMEC/D3 cells (Zhong et al, 2008).
RhoA GTPase Pull Down Assay
RhoA activity was assessed by the RhoA activation assay kit according to the manufacturer's instruction (Millipore). Briefly, treated cells were washed twice with ice-cold PBS and lysed in a buffer containing 25 mmol/L HEPES (pH 7.5), 150 mmol/L NaCl, 1% lgepal CA-630, 10 mmol/L MgCl2, 1 mmol/L EDTA, 10% glycerol, 10 μg/ml aprotinin, and 10 μg/ml leupeptin. The extracts were centrifuged at 13,000
Western Blotting
Treated cells were fixed with ice cold 10% trichloroacetic acid containing 10 mmol/L dithiothreitol (DTT) and immediately frozen in liquid nitrogen. Before analysis the cells were thawed, scraped out, and centrifuged at 14,000
Adenovirus Infection
We constructed adenovirus encoding for the Rho inhibitor C3 transferase, the exoenzyme that specifically ribosylates and inhibits the function of Rho gene. This adenovirus (termed C3 adenovirus) expresses C3 transferase under the control of the CMV promoter with green fluorescent protein (GFP) as a separate tracer. As GFP gene is downstream from an internal ribosome entry site element, C3 transferase and GFP are expressed from the same transcript. A separate adenovirus vector expressing GFP without exoenzyme C3 transferase was also constructed and served as a control. The viruses were amplified by repeated infection of human embryonic kidney (HEK) 293 cells. The media containing recombinant adenoviruses were collected and stored at −80°C.
hCMEC/D3 cultures at 50% to 60% confluence were subjected to infection with recombinant adenoviruses at a multiplicity of infection of 200. The infected cells were used for experiments 2 days postinfection when the cultures reached full confluence. Recombinant molecules were expressed in ∼100°/o hCMEC/D3 cells as monitored by detection of GFP signal under a Nikon fluorescent microscope.
Rhodamine 123 Efflux Assay
The functional activity of the P-gp efflux transport system was evaluated as described earlier (Hayashi et al, 2005). Briefly, confluent hCMEC/D3 cells were treated with Tat (100 nmol/L) for 15 h in the presence or absence of ROCK inhibitor Y27632 (5 μmol/L). The media were removed, the cells were washed and incubated with rhodamine 123 (10 μmol/L) dissolved in Krebs-Ringer glucose buffer for 1 h in 37°C in cell culture incubator. The reaction was terminated by removing rhodamine 123 solution and washing the cells with ice-cold PBS. The cells were solubilized with NaOH, neutralized with HCl, and cellular levels of rhodamine 123 were determined by fluorescent measurement (excitation, 505 nm and emission, 534 nm). The cellular content of rhodamine 123 was quantified according to a standard curve and normalized per cellular protein content.
Promoter Activity Assay
Multidrug resistance gene promoter construct linked to chloramphenicol acetyltransferase (MDR-1-CAT) was kindly provided by Dr Gottesman (NCI, Bethesda, MD, USA). Chloramphenicol acetyltransferase activity in cell extracts was measured by CAT ELISA kit (Roche, Mannheim, Germany) following the manufacturer's instruction. Briefly, hCMEC/D3 cells were seeded on 6-cm dishes and transfected on the next day with 4 μg of plasmid DNA using the Lipofectin Reagent (Invitrogen, Carlsbad, CA, USA). The cells were starved for additional day and treated with 100 nmol/L Tat for 1.5 h. Protein concentrations were determined by Coomassie Bradford protein assay kit (Pierce), and 100 μg protein from each sample was used for CAT assay.
Cholesterol Depletion and Caveolin-1 Silencing
Depletion of cellular cholesterol was achieved by treatment of hCMEC/D3 cells with 1 mmol/L MβCD for 1 h at 37°C. Caveolin-1 silencing was performed as described earlier (Zhong et al, 2008), using a mixture of siRNA corresponding to the nucleotides 69 to 87 of the human caveolin-1 mRNA sequence: 5′-CAUCUACAAGCCCAACAAC-dTdT-3′ (cav-1 siRNA-1) and to the nucleotides 223 to 241: 5′-CCA GAAGGGACACACAGUU-dTdT-3′ (cav-1 siRNA-2). Control siRNA was: 5′-AAAGAGCGACUUUACACAC-dTdT-3′. LipofectAMINE-plus (Invitrogen) was used to transfect hCMEC/D3 overnight with cav-1 siRNA-1 (40 nmol/L), followed by transfection with cav-1 siRNA-2 (40 nmol/L) for additional 5 h.
Statistical Analysis
Each experiment was repeated a minimum of three times. One-way or two-way ANOVA was used to compare mean responses among the treatments. Statistical probability of
Results
Tat Stimulates RhoA Activation in Brain Endothelial Cells
Tat can interact with cell surface receptors, such as vascular endothelial growth factor receptor type 2 (VEGFR-2) (Andras et al, 2005), and thus activate small GTPases. In addition, the Rho pathway was reported to be activated in response to exposure to HIV (Persidsky et al, 2006). Therefore, we studied activation of RhoA in hCMEC/D3 cells exposed to HIV protein Tat. RhoA stimulation is connected to the transformation of GDP-RhoA to GTP-RhoA. As illustrated in Figure 1A, exposure to 100 nmol/L Tat resulted in a rapid and time-dependent increase in GTP-RhoA. GTP-RhoA levels were elevated in cells treated with Tat for 3 or 10 mins, returning to the control values in cultures exposed to Tat for 30 mins or longer. The total RhoA level was not affected by Tat exposure. In addition, treatment with immunoabsorbed Tat (AA-Tat, negative control) did not alter GTP-RhoA levels. The effects of Tat on RhoA activation were dose-dependent and the highest levels of GTP-RhoA (∼ 3-fold increase over basal values) were observed in cells exposed to Tat at the concentration of 100 nmol/L (Figure 1B).
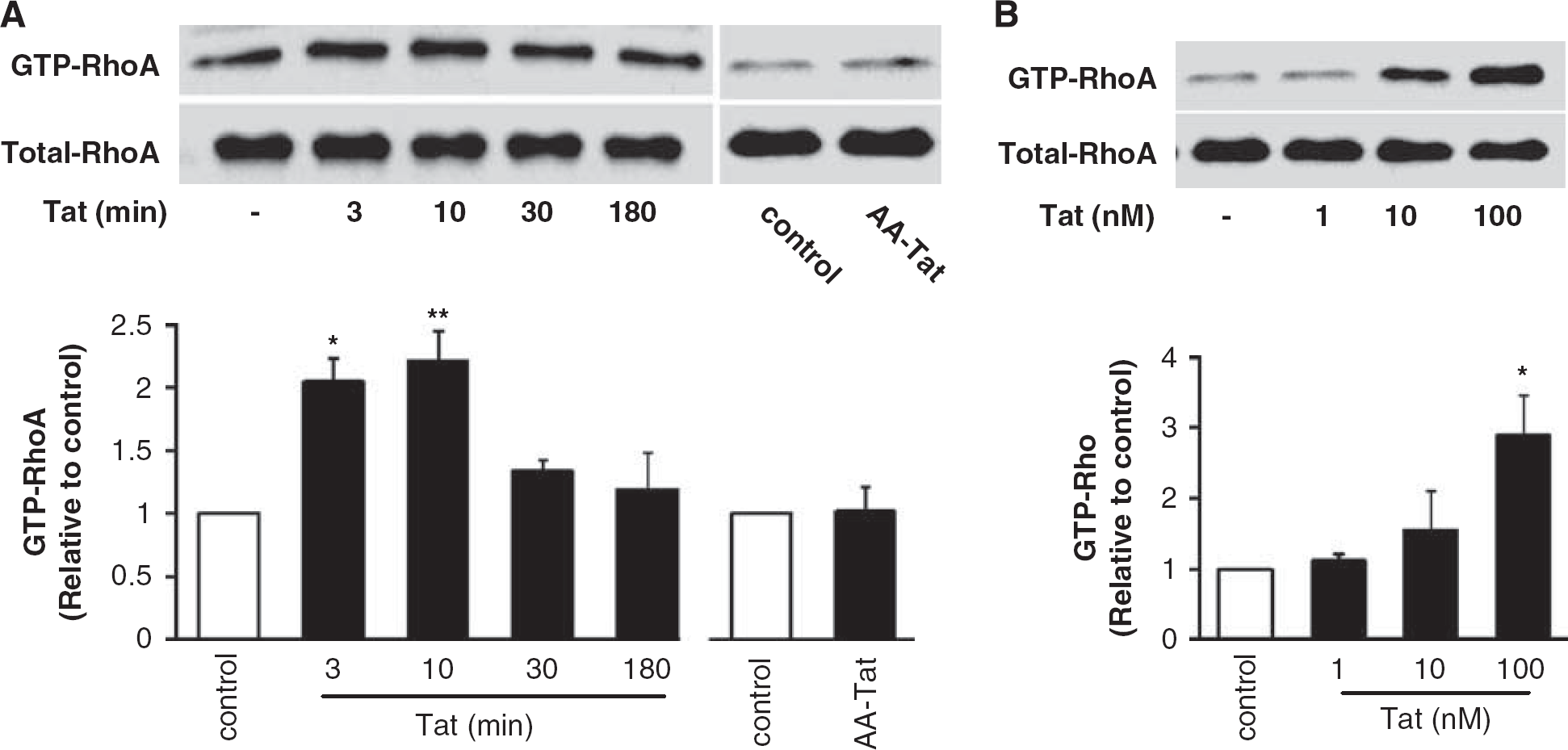
Exposure to Tat activates RhoA in hCMEC/D3 cells. (
Tat Activates Downstream Kinases of the RhoA Signaling Cascade
The biological effects of Rho are mediated by a number of downstream effector proteins, including RhoA/Rho kinase (ROCK). Activation of the ROCK pathway may result in inhibition of MLCP and maintaining MLC in the phosphorylated status (Rolfe et al, 2005). To determine whether these kinases provide an active signaling pathway in Tat-induced stimulation of brain endothelial cells, we first evaluated the levels of phosphorylated myosin phosphatase target subunit 1 (p-MYPT-1), an integral component of MLCP in hCMEC/D3 cells treated with 100 nmol/L Tat for up to 1 h. As shown in Figure 2A, exposure to Tat resulted in an increase in phosphorylation of MYPT-1 with a peak at 15 mins (∼ 2-fold increase), followed by a decline to control values 30 mins posttreatment. Exposure to AA-Tat did not have any effect on MYPT-1 phosphorylation. To further confirm specificity of these responses, hCMEC/D3 cells were pretreated with Y27632 (5 μmol/L), a specific inhibitor of ROCK, followed by exposure to Tat for 15 mins. Figure 2B indicates that preincubation with Y27632 efficiently blocked Tat-stimulated MYPT-1 phosphorylation without altering the total MYPT-1 levels.
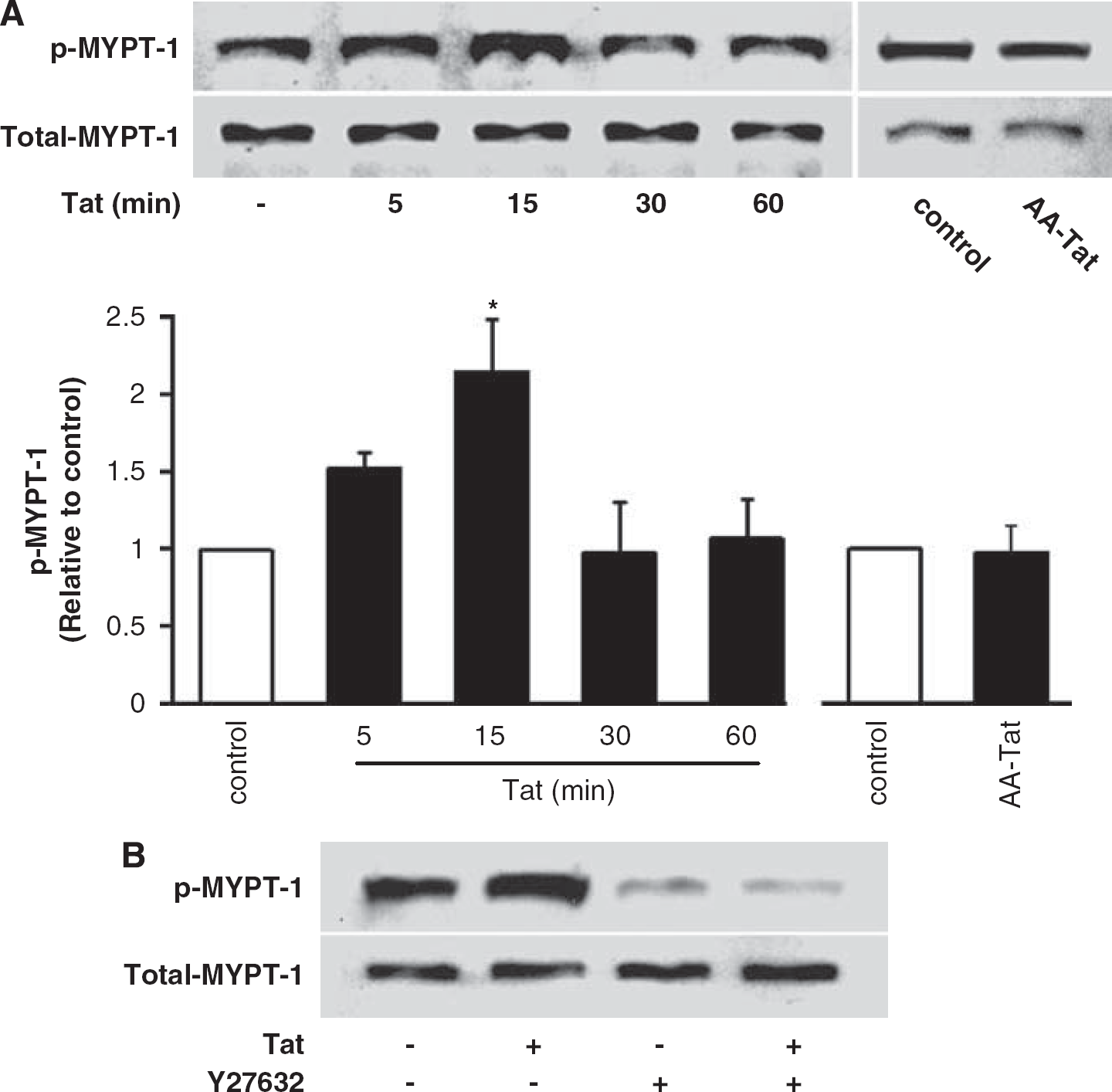
Tat activates myosin phosphatase target subunit 1 (MYPT-1) via the Rho/ROCK pathway. (
Phosphorylation of MYPT-1 affects the ability of MLCP to dephosphorylate MLC, allowing MLC to retain its phosphorylated status. Indeed, treatment with Tat but not AA-Tat significantly upregulated levels of phosphorylated 20-kDa MLC (∼1.9-fold increase over basal levels) without changes in the total levels of this protein (Figure 3A). These effects were the most pronounced in cells exposed to Tat for 1 h. Tat-induced activation of MLC was markedly blocked by the ROCK inhibitor Y27632 (Figure 3B) and MLCK inhibitor ML-9 (10 μmol/L) (Figure 3C), indicating that Tat-induced activation of MLC is a downstream effect of ROCK stimulation.
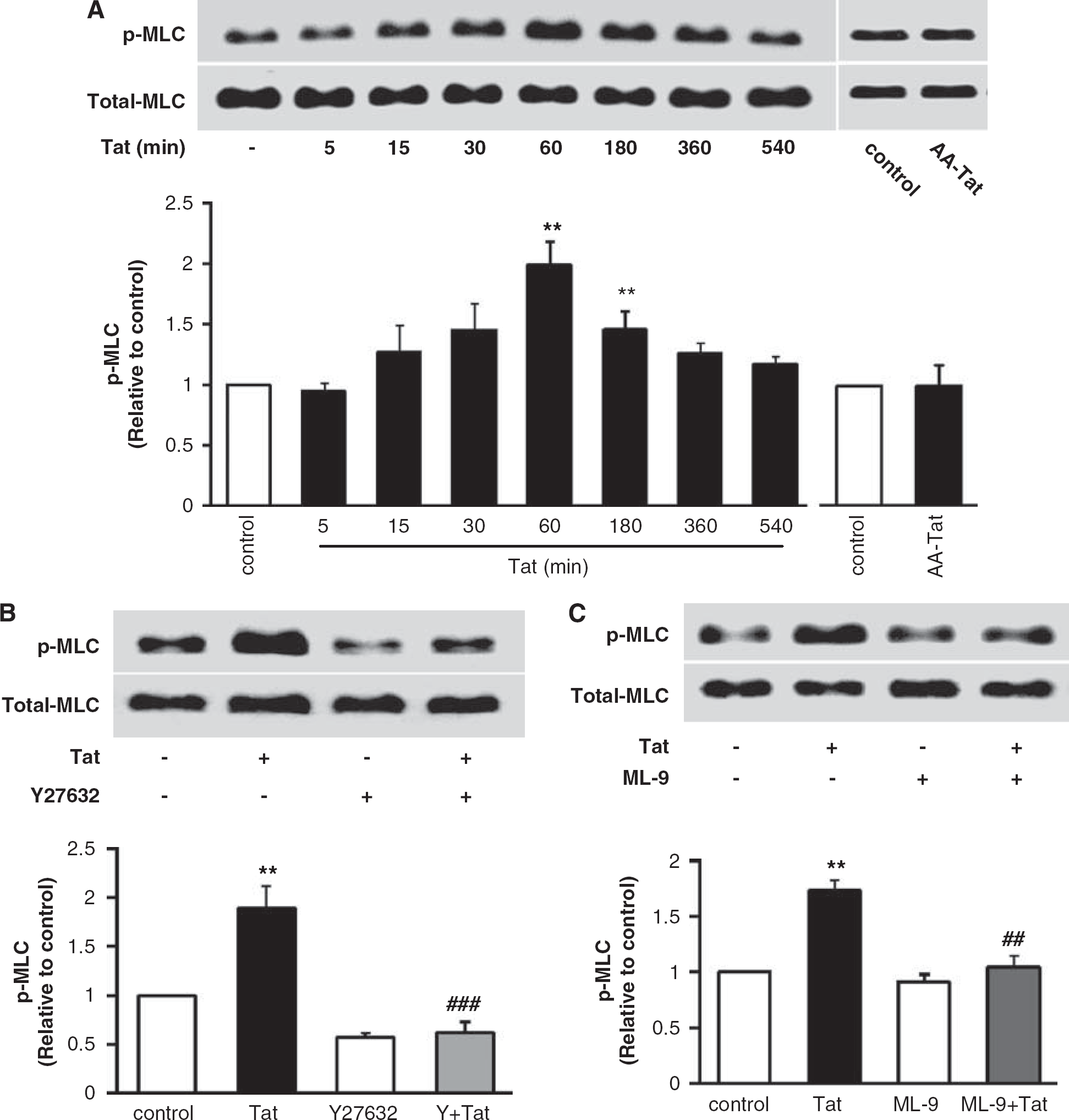
The Rho/ROCK signaling is involved in Tat-induced phosphorylation of myosin light chain (MLC). (
Tat Upregulates P-glycoprotein Expression via the Rho-Mediated Signaling
In search of potential targets of Rho signaling, we focused on P-gp protein expression as the principal efflux transporter in brain endothelial cells. Treatment of confluent hCMEC/D3 cultures with Tat resulted in a dose- and time-dependent increase in P-gp levels. The most pronounced increase in P-gp level was observed after a 15-h exposure to Tat (Figure 4A). The specificity of this effect was confirmed by treatment with AA-Tat. In dose-dependent experiments, Tat at 10 nmol/L, and in particular at 100 nmol/L, resulted in effective elevation of P-gp protein levels with the maximal induction reaching an ∼2-fold increase over basal level (Figure 4A). Actin was determined as a housekeeping protein and its levels were not changed in response to Tat or AA-Tat.
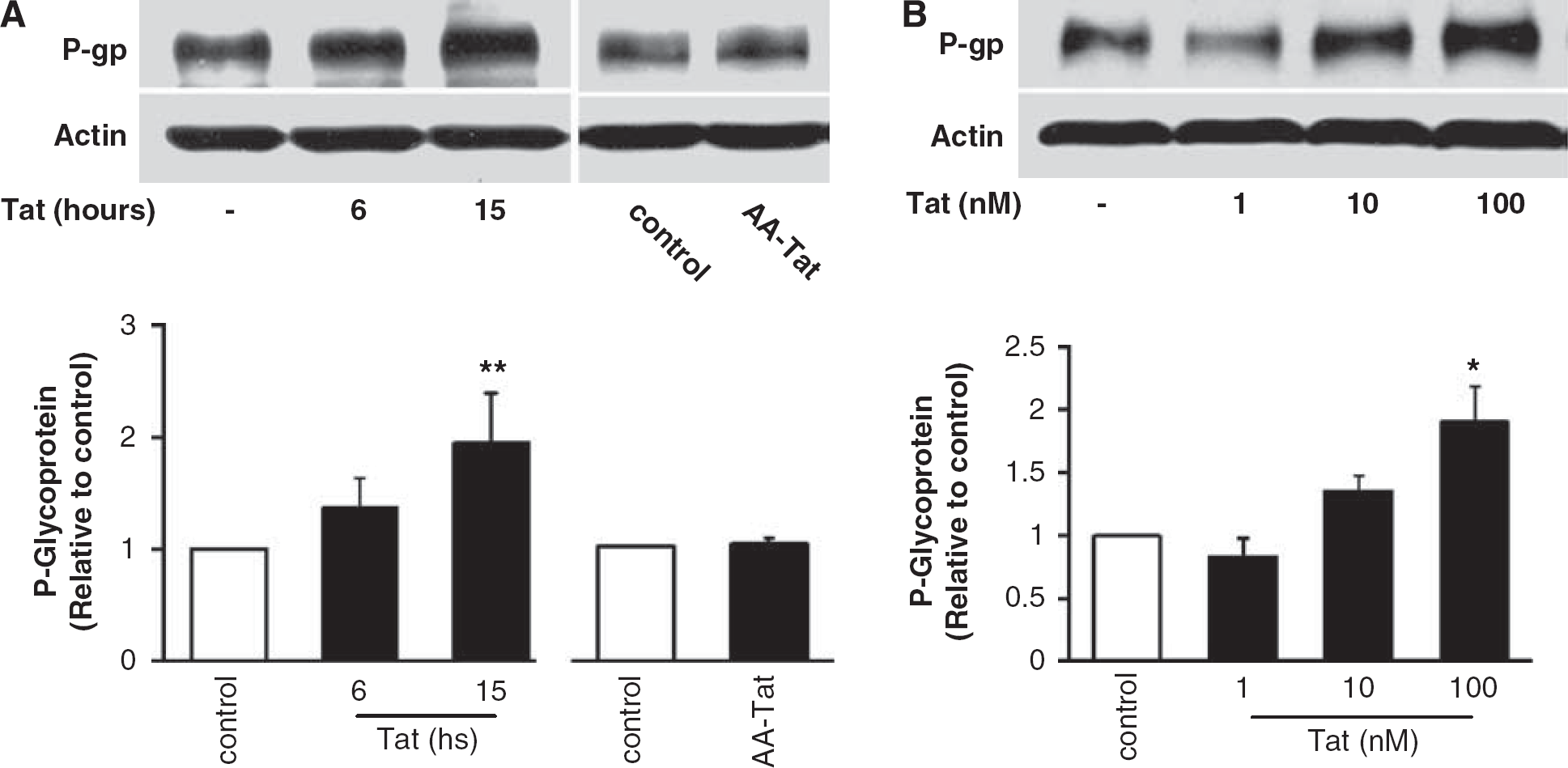
Tat induces P-gp upregulation in brain endothelial cells.
To evaluate the involvement of Rho signaling in Tat-induced overexpression of P-gp, Rho activity was blocked by infecting endothelial cells with an adenovirus expressing Rho inhibitor, exoenzyme C3 transferase (C3-adenovirus). Control cells were mock-infected or infected with the control adenovirus. The cultures were then treated with Tat for 15 h for determination of P-gp protein expression. To evaluate MDR-1 promoter activity, hCMEC/D3 cultures were transfected with pMDR-1-CAT construct, followed by exposure to Tat (100 nmol/L) for 1.5 h. As indicated in Figure 5A (left panel), treatment with Tat significantly increased activity of MDR-1 promoter in cultures mock-infected or infected with a control adenovirus. However, inhibition of Rho by C3-adenovirus markedly protected against Tat-induced MDR-1 activity. These effects were fully correlated with the protective effects of C3-adenovirus on Tat-induced upregulation of P-gp protein levels (Figure 5A, right panel). The observed inhibition by C3 adenovirus was specific because infection with control adenovirus alone did not affect the basal level of P-gp protein expression or actin levels.
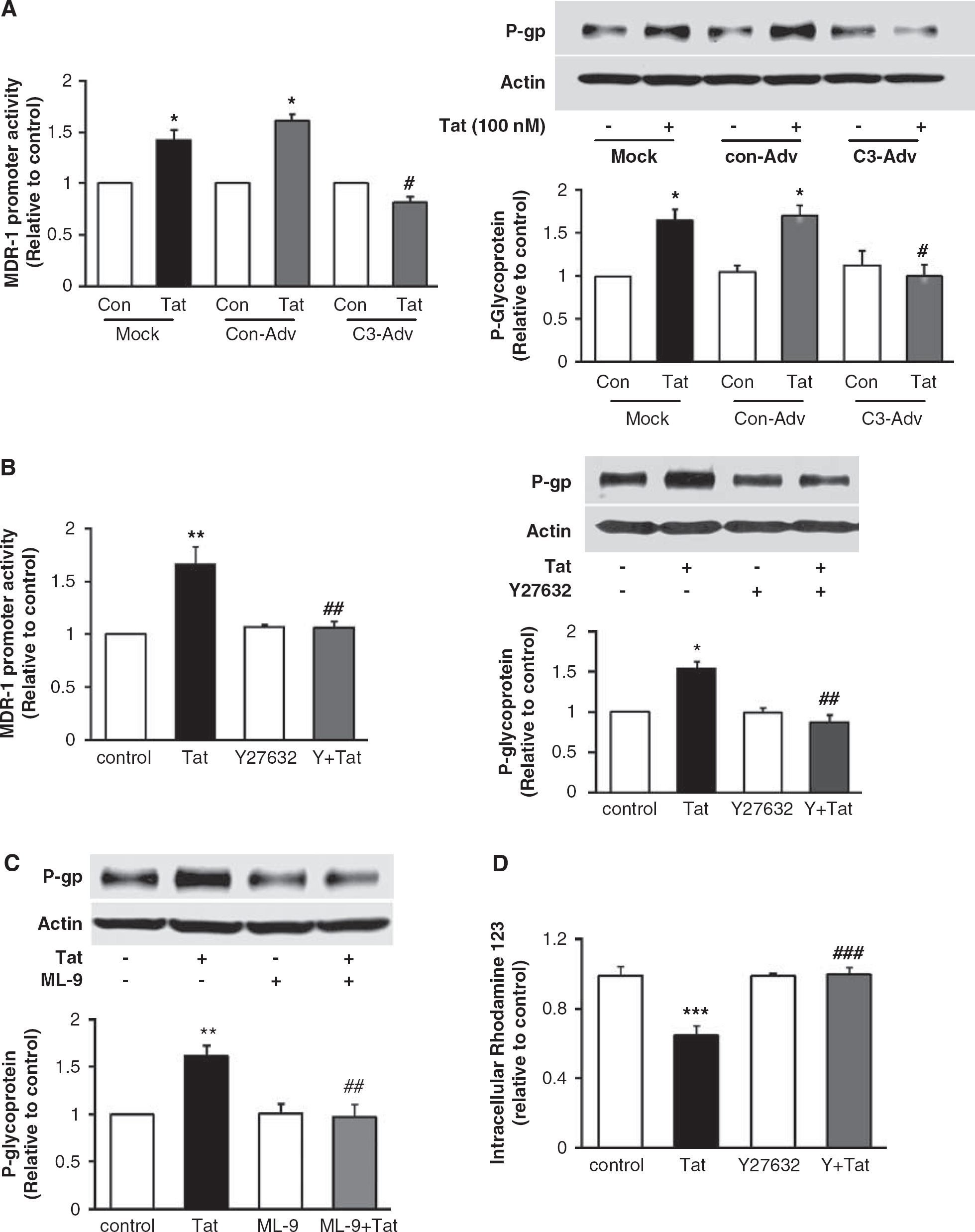
Inhibition of the Rho/ROCK pathway protects against Tat-induced upregulation of P-gp at the transcriptional level. (
To further determine the relationship between the Rho signaling on P-gp expression, we next inhibited ROCK activity by a 30-min pretreatment with Y27632 (5 μmol/L). In addition, selected cultures were transfected with the MDR-1-CAT promoter construct. Similar to C3 adenovirus, treatment with Y27632 markedly protected against Tat-induced upregulation of MDR-1 promoter activity and P-gp protein levels (Figure 5B, left and right panels, respectively). In addition, a 30-min pretreatment with ML-9 (10 μmol/L) significantly protected against Tat-mediated overexpression of P-gp protein without affecting its basal levels (Figure 5C).
Tat-induced P-gp levels were correlated with increased efflux activity. As shown in Figure 5D, treatment with Tat (100 nmol/L) significantly decreased cellular accumulation of rhodamine 123, indicating an accelerated removal of this dye. Importantly, ROCK inhibitor Y27632 fully abolished these effects.
Intact Lipid Rafts are Critical for Tat-Induced P-glycoprotein Upregulation and RhoA Activation
P-glycoprotein is localized at the luminal membranes of endothelial cells; however, controversy exists regarding its association with lipid rafts and/or caveolae. Therefore, in the last series of experiments, we focused on evaluation of the function of lipid rafts or functional caveolae in Tat-induced upregulation of P-gp. Depletion of membrane cholesterol by exposing the cells to 1 mmol/L MβCD for 1 h was used to disrupt lipid rafts. As indicated in Figure 6A, cholesterol depletion markedly protected against Tat-induced activation of the RhoA signaling. Most importantly, this procedure was also effective in blocking Tat-induced upregulation of P-gp (Figure 6B). Treatment with MβCD did not alter the basal levels of GTP-RhoA or P-gp. In contrast, silencing caveolin-1 expression did not block Tat-mediated activation of RhoA or overexpression of P-gp (Figures 6C and 6D, respectively) in hCMEC/D3 cells.
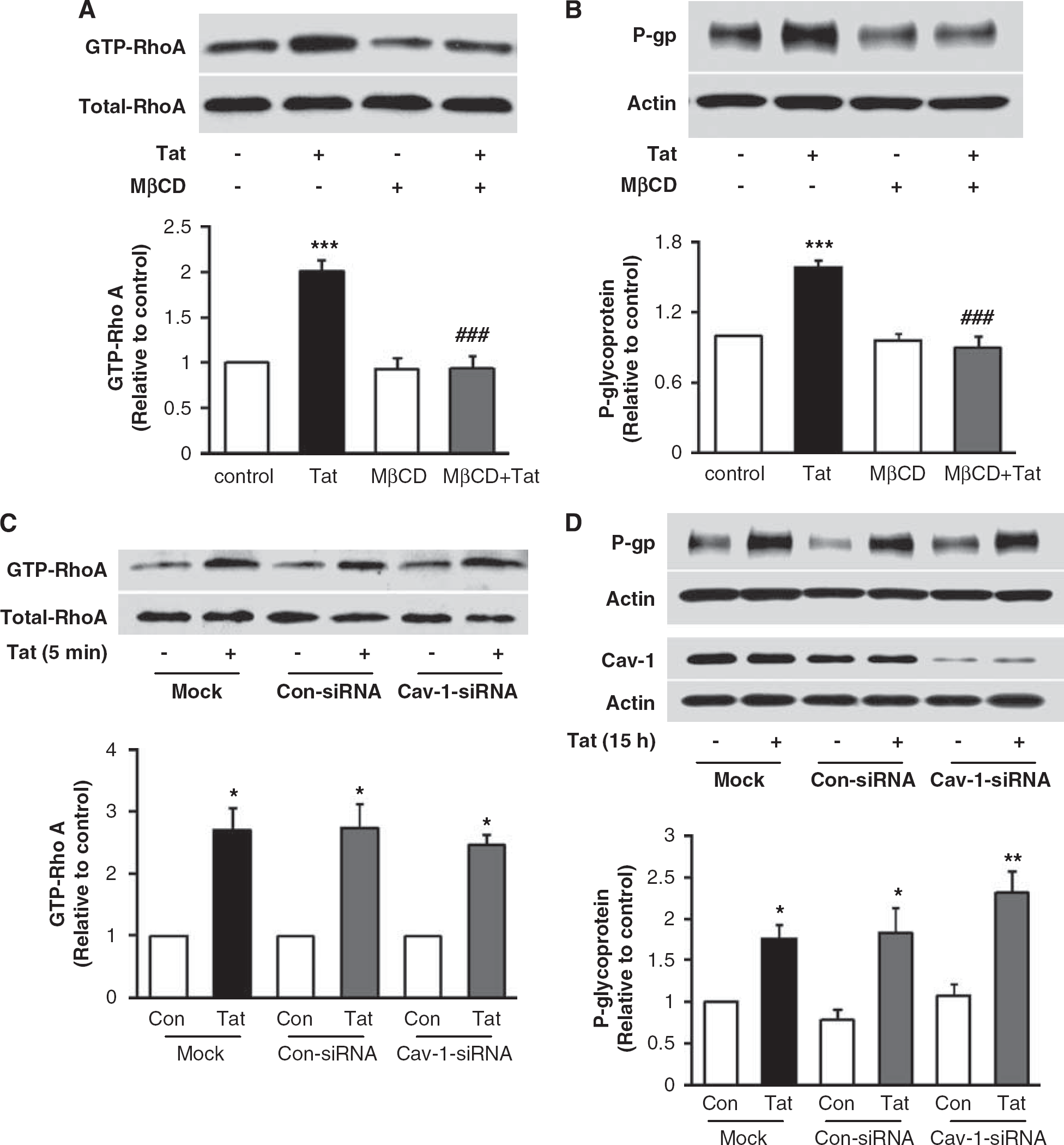
Lipid rafts but not caveolae modulate Tat-induced upregulation of P-gp expression in brain endothelial cells. Depletion of membrane cholesterol protects both against Tat-induced activation of RhoA (
Discussion
Successful treatment of HIV necessitates suppressing of viral replication throughout the body compartments. This is of particular importance in the CNS because the BBB restrains the delivery of antiviral drugs into the brain. A key component of the BBB that limits drug availability in the brain is the presence of a complex system of drug efflux transporters that remove a broad range of compounds from the brain. These transporters include the members of the ATP-binding cassette superfamily of proteins, such as P-gp, breast cancer resistance protein, and multidrug resistance proteins. Among them, P-gp has the primary impact on the penetration of several classes of drugs into the brain. To a large degree, activity of P-gp determines the clinical usefulness, side effects, and toxicity risks of drugs. In the context of HAART, it is important to note that P-gp limits brain entry of HIV-1 protease inhibitors. Thus, the ability of these drugs to achieve therapeutic concentrations in the brain is limited, thereby creating a potential sanctuary for viral replication (Miller et al, 2008; Ronaldson et al, 2008; Spitzenberger et al, 2007).
Both viral factors and drugs used for HAART may be responsible for upregulation of P-gp in HIV-1-infected patients on antiretroviral therapy. Indeed, protease inhibitors are not only substrates for P-gp but they also can induce overexpression of P-gp via a process that is at least partially mediated by the nuclear receptor, pregnane X receptor (Ott et al, 2009). These effects were demonstrated in several different cell types, including brain endothelial cells (Zastre et al, 2009). In addition, HIV-1 and HIV-1 specific proteins may contribute to upregulation of P-gp. A relatively recent study based on brain autopsy samples of HIV patients reported a correlation between P-gp expression and HIV RNA levels (Langford et al, 2004). To support these findings, it was shown that HIV infection
The importance of P-gp upregulation in preventing the effective delivery of many drugs into the CNS necessities studies on regulatory mechanisms that may be involved in this process. The signaling mechanisms leading to overexpression of P-gp are complex; however, most of the published evidence suggests an important function of oxidative stress and redox-regulated pathways in P-gp upregulation. For example, it was demonstrated that induction of NADPH oxidase with the subsequent activation of the JNK/c-Jun signaling is involved in overexpression of P-gp (Hartz et al, 2008). Studies indicated that endothelin-1 acting via the ETB receptor and nuclear-factor-κB can lead to P-gp overexpression (Bauer et al, 2007). These results are in agreement with our observations that blocking nuclear-factor-κB by SN50 prevents Tat-induced overexpression of P-gp in primary cultures of brain endothelial cells (Hayashi et al, 2005). In addition, the function of ERK1/2 pathway and β-catenin in upregulation of P-gp were reported (Lim et al, 2008). However, there are no earlier reports on the function of the Rho signaling pathway in P-gp expression.
The RhoA/ROCK signaling pathway has been shown to contribute to several processes that are critical for the integrity of the brain endothelium. When bound to GTP, Rho interacts with ROCK to affect actin organization. Actin–myosin interaction and loss of junctional integrity are pivotal processes in disruption of tight junction integrity and increased endothelial permeability. Indeed, activation of the RhoA/ROCK signaling has been implicated in increased transendothelial leukocyte migration (Persidsky et al, 2006). Organization of actin filaments can also influence P-gp expression and the development of drug resistance. It has been reported that P-gp can interact with actin cytoskeleton through ezrin, radixin, and moesin proteins and that this interaction results in P-gp delocalization and alterations of P-gp efflux function (Luciani et al, 2002). It was also observed that inhibition of actin polymerization can lead to the loss of P-gp function in P-gp-overexpressing multidrug resistant osteosarcoma cells (Takeshita et al, 1998). In line with these reports, our data indicate that Tat-mediated activation of the RhoA/ROCK signaling can sequence to phosphorylated MLC, which is effective in cross-linking of F-actin into stress fibers. On the other hand, activation of the Rho pathway can upregulate P-gp expression at the transcriptional levels as evidenced in this study by MDR-1 mRNA overexpression and induction of MDR-1 promoter activity in Tat-treated hCMEC/D3 cells.
Biological effects of Tat are mediated by membrane mechanisms and cell surface receptors located in lipid rafts and caveolae. For example, Tat can induce the Ras/ERK1/2 signaling and disrupt claudin-5 expression through activation of lipid raft-localized VEGFR-2 (Andras et al, 2005). Interestingly, silencing of caveolin-1 protected against Tat-induced activation of the Ras signaling and tight junction disruption (Zhong et al, 2008). Therefore, we evaluated the involvement of caveolae and lipid rafts in Tat-induced upregulation of P-gp. This focus was also supported by the observation that P-gp expression and activity can be regulated by cellular cholesterol and caveolin-1 expression (Orlowski et al, 2006).
Caveolin-1 silencing experiments performed in this study did not inhibit Tat-induced activation of Rho or P-gp expression. These results were unexpected because earlier experiments indicated interrelationship between caveolin-1 and P-gp expression in MDR cells (Zhu et al, 2004). In addition, it was reported that P-gp can interact with caveolin-1 and caveolin-2 located in caveolae and that caveolin-1 can inhibit transport activity of P-gp in bovine brain endothelial cells (Jodoin et al, 2003). However, different cell types used for these experiments may account for the different results. Although the majority of data in literature indicates the localization of P-gp in caveolae (Rolfe et al, 2005; Ronaldson et al, 2004), other reports suggest lipid rafts but non-caveolar localization of this efflux transporter (Radeva et al, 2005) or localization both in caveolae and non-caveolae membrane domains (Jodoin et al, 2003).
Lipid rafts are membrane microdomains that are enriched with cholesterol; thus, cholesterol depletion by MβCD is an effective strategy to disrupt lipid rafts. In this study, exposure to 1 mM MβCD for 1 h completely protected against Tat-induced activation of the Rho signaling and P-gp overexpression. Such results are in agreement with earlier findings demonstrating that cholesterol depletion from the cell membrane of Caco-2 cells induced a decrease in binding of saquinavir (Asawakarn et al, 2001). Importantly, repletion of cholesterol in MβCD-treated cells restored P-gp transport activity (Jodoin et al, 2003; Luker et al, 2000).
In conclusion, this study demonstrates that exposure to Tat results in activation of the Rho signaling and upregulation of P-gp protein expression in brain endothelial cells. These events are interrelated, because inhibition of the Rho pathway prevents Tat-induced upregulation of P-gp. In addition, intact lipid rafts but not caveolae are required for Tat-mediated activation of the Rho signaling and P-gp overexpression (Figure 7). These results suggest that Tat-induced Rho activation may constitute an early signaling mechanism leading to upregulation of efflux transporters on the brain endothelium thereby limiting antiretroviral drug penetration into the CNS.
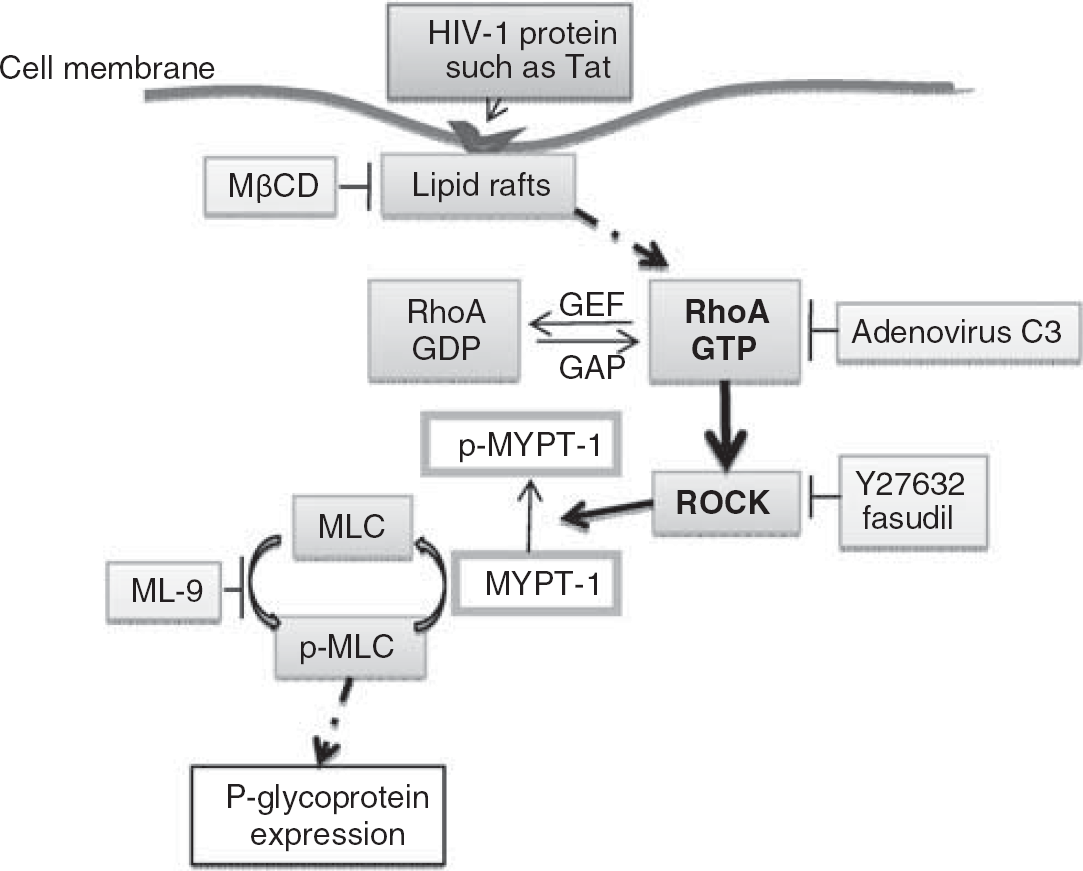
The proposed signaling pathway of Tat-induced upregulation of P-gp expression in human brain endothelial cells. The HIV-1 protein Tat, acting via intact lipid rafts, activates RhoA and ROCK kinases, leading to inhibition of MLCP (via phosphorylation of MYPT-1) and increased phosphorylation of MLC. This study provides novel evidence that these events are involved in the regulation of P-gp expression.
Footnotes
Acknowledgements
We greatly thank Dr Michael M Gottesman (Laboratory of Molecular Biology, National Cancer Institute, Bethesda) for providing the MDR-1-CAT construct. We also thank Dr Pierre-Olivier Couraud (Institut COCHIN, Paris, France) for providing the hCMEC/D3 cells. This work was supported by MH063022, MH072567, and NS39254. Generation of Tat protein was supported by P01 DA19398.
The authors declare no conflict of interest.