
Abstract
Cerebral blood flow (CBF) and cerebral metabolic rate are normally coupled, that is an increase in metabolic demand will lead to an increase in flow. However, during functional activation, CBF and glucose metabolism remain coupled as they increase in proportion, whereas oxygen metabolism only increases to a minor degree—the so-called uncoupling of CBF and oxidative metabolism. Several studies have dealt with these issues, and theories have been forwarded regarding the underlying mechanisms. Some reports have speculated about the existence of a potentially deficient oxygen supply to the tissue most distant from the capillaries, whereas other studies point to a shift toward a higher degree of non-oxidative glucose consumption during activation. In this review, we argue that the key mechanism responsible for the regional CBF (rCBF) increase during functional activation is a tight coupling between rCBF and glucose metabolism. We assert that uncoupling of rCBF and oxidative metabolism is a consequence of a less pronounced increase in oxygen consumption. On the basis of earlier studies, we take into consideration the functional recruitment of capillaries and attempt to accommodate the cerebral tissue's increased demand for glucose supply during neural activation with recent evidence supporting a key function for astrocytes in rCBF regulation.
Keywords
Introduction
More than 100 years ago, Roy and Sherrington (1890) came to the conclusion that the vascular supply of the brain can be varied locally in correspondence with local variations in functional activity. In the early 1960s, Lassen and Ingvar introduced a methodology for measurement of regional cerebral blood flow (rCBF) in humans, and during the 1970s, once it was more widely acknowledged that rCBF increases in response to cerebral activation, these methods were applied to functional brain mapping (Lassen et al, 1978). Although it had already been known for a while that cortical oxygen tension increases in response to activation (Cooper et al, 1966), it was only somewhat later realized that regional cerebral oxygen consumption does not increase to the same extent as does rCBF and rCMRglc (Fox et al, 1988). This important physiologic phenomenon, commonly referred to as the
The cause of the rCBF increase during activation is still not entirely clear. Some authors have argued that, because of diffusion limitations, there will be limitations in oxygen availability in the tissue and no reserve pool. For this reason, the rCBF increases must always be disproportionate to changes in oxidative metabolism, to the extent that the increases in capillary PO2 needed to support an increased oxidative metabolism under these conditions can only be achieved through a large flow increase (Gjedde, 1991; Buxton and Frank, 1997). In contrast with the excess increase in rCBF compared with oxygen consumption, there is a
Capillary heterogeneity
As the concept of capillary heterogeneity is of relevance for discussion of both glucose and oxygen transport in the brain, a brief overview will be given here. Recruitment of capillaries with opening of closed non-perfused capillaries, as it takes place in muscles during exercise, will increase the effective capillary surface area and thereby the flux of substances across the capillary as long as permeability remains unchanged. A functional recruitment with slowly perfused capillaries becoming rapidly perfused may be a physiologic phenomenon of significant importance for substrate transport across the BBB (Hertz and Paulson, 1980; Knudsen et al, 1990; Kuschinsky and Paulson, 1992). Thus, in baseline conditions, the cerebral microcirculation is relatively heterogeneous, but it becomes gradually more homogeneous as CBF increases (Kuschinsky and Paulson, 1992).
The notion of capillary heterogeneity in the brain is strongly supported by studies of direct microscopic observation of the cerebral microcirculation (Pawlik et al, 1981) and
Still the question of capillary recruitment is often considered controversial (Buxton, 2002) and other mechanisms have been or are still debated, for example increased velocity without recruitment.
Glucose consumption—cerebral blood flow coupling
Earlier studies conducted in humans in resting state have found a quite consistent ratio of about 5.6 between glucose and oxygen brain uptake (Madsen et al, 1995; Hasselbalch et al, 1996). These studies suggest that glucose is almost completely oxidized to CO2 and H2O through glycolysis and the subsequent tricarboxylic acid (TCA) cycle. Glucose consumption seems tightly coupled to neurotransmitter recycling and restoration of neuronal membrane potentials through conversion of glucose to lactate in astrocytes, and shuttling of lactate to neurons for oxidation (Rothman et al, 1999; Magistretti and Pellerin, 1999; Pellerin and Magistretti, 2004; Pellerin et al, 2007). Thus, in the resting state, coupling of CMRglc and neural activity seems to be regulated by the need for energy substrates to maintain synaptic transmission.
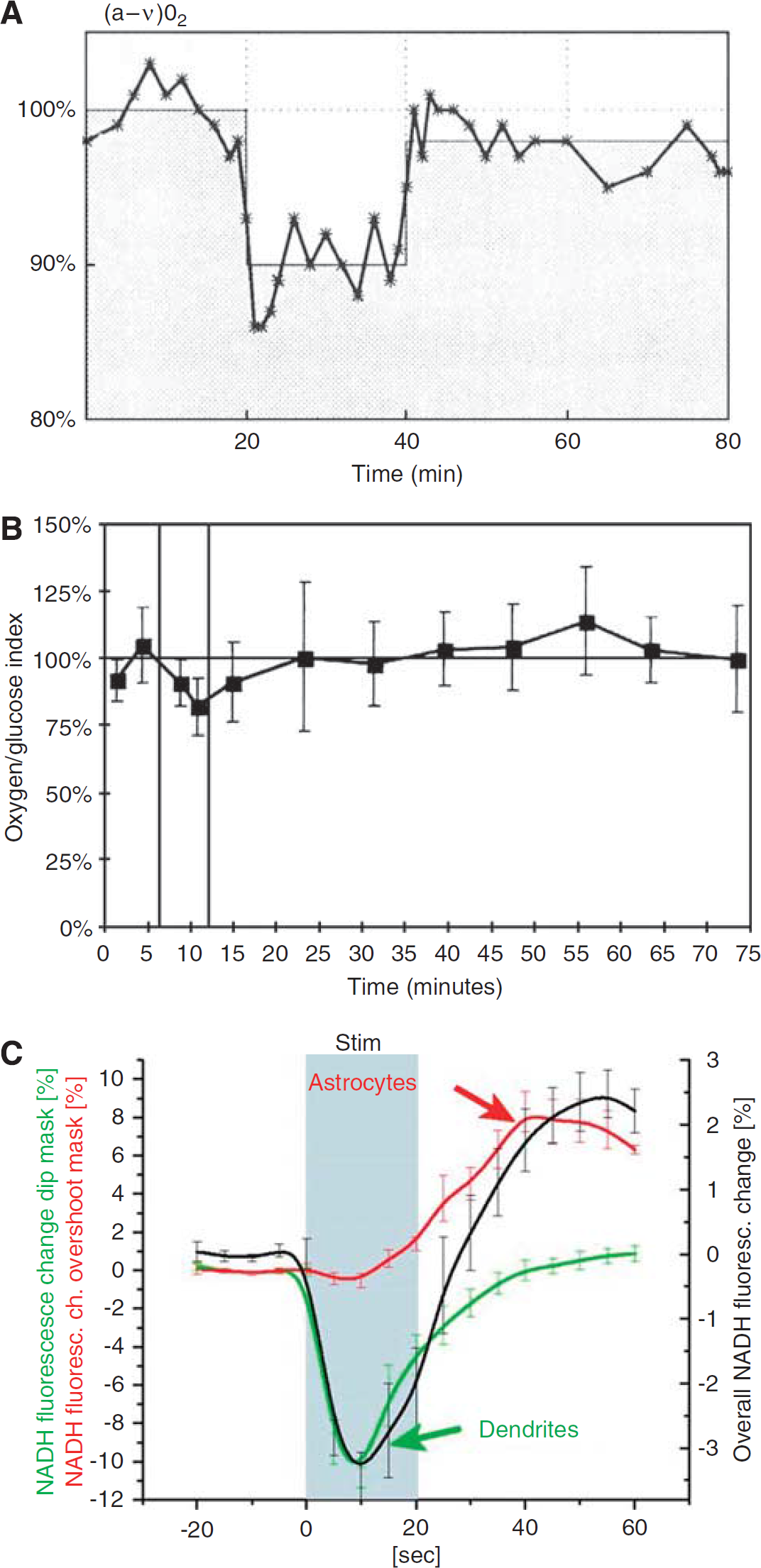
Further, a tight coupling between CBF and glucose consumption during functional activation was established by several earlier studies. During epileptic seizure, both CBF and glucose uptake increase by at least a factor of 2 (Brodersen et al, 1973). In studies of functional activation in humans, parallel increases in CBF and glucose consumption have been found (Fox et al, 1988; Madsen et al, 1995; Newberg et al, 2005) (Figure 1A). In addition, in experimental studies in rats, the CBF increase correlated better to the increase in glucose uptake than to the increase in oxygen uptake, although the CBF increase was more marked (Madsen et al, 1998). However, the exact coupling (% increase) of flow to the glucose consumption seen in the human studies was not observed in the experimental study in which flow increased by 68% and glucose consumption only by 35%, as the (a-v)glu dropped by 20%. We consider this to reflect a statistical variation and not a biological phenomenon. Thus, in a subsequent study using exactly the same activation paradigms in the same laboratory, a drop in (a-v)glu could not be confirmed, but in contrast, an increase by 13% was observed.
(
During activation, the oxygen/glucose uptake ratio decreased by 20–25% (Madsen et al, 1998; Madsen et al, 1999) indicating that the excess CBF and glucose uptake during activation is not used for increased oxidative glucose metabolism (Figure 1B). Correspondingly, there is a modest increase in brain lactate concentration in the activated areas (Prichard et al, 1991; Sappey-Marinier et al, 1992; Frahm et al, 1996; Urrila et al, 2003; Mangia et al, 2007), and an increase in extracellular lactate concentration has been observed during functional activation (Hu and Wilson, 1997). Furthermore, a slight excess lactate efflux from the brain takes place during activation (Madsen et al, 1999). But oxidative metabolism accounts only for 60% of the glucose taken up by the brain, and of the non-oxidized glucose, only ∼50% seems to be metabolized to lactate escaping from the brain (Madsen et al, 1999). Further, by increasing blood lactate levels during maximal exercise in humans, lactate influx increases, but lactate seems not to be oxidized during the experimental time (Dalsgaard et al, 2004). All these early studies would, at first glance, seem to be compatible with the notion that once taken up by astrocytes, glucose is converted to lactate without being oxidized—at least not during the experimental time. More recent data support the astrocyte–neuron lactate shuttle hypothesis: neurons and astrocytes have comparable uptakes of glucose (Nehlig et al, 2004), but 90–95% of the energy coming from the glucose uptake is consumed by neurons (Attwell and Laughlin, 2001), suggesting that astrocytes deliver glucose (mostly in the form of lactate) to neurons for energy production. Further, neurons have a preference for metabolizing lactate over glucose (Smith et al, 2003). However, as covered in a recent review (Hertz et al, 2007), these concepts are open to challenge. As described in the section on the astrocyte–arteriole connection, the tight coupling between rCBF and rCMRglc during activation seems well established, and earlier data support the idea that lactate, whether produced peripherally and taken up by the brain, or produced by astrocytes, is accumulating in the brain during activation. Further, as brain lactate concentration and oxygen consumption do not increase markedly during activation, lactate must be metabolized by other pathways than through the TCA cycle. In a recent elegant study, Cruz et al (2007) used labeled deoxyglucose and glucose during functional activation to study local glucose uptake and spreading of labeled glucose metabolites, respectively. They found that during rest and activation, glucose metabolites including lactate, had a spatial spread 1.5–2.2-fold larger than deoxyglucose, and the tissue volume labeled by glucose metabolites increased 20–50% during activation compared with the resting condition. These results may explain the local and temporary decrease in the oxygen to glucose metabolic ratio. Thus, glucose metabolites are quickly being dispersed from the activated area, and subsequently, either released from the brain, entering metabolite pools or oxidized in the ‘recovering phase’ of activation, as suggested by an increase in the oxygen/glucose metabolic ratio after activation (Madsen et al, 1998; Madsen et al, 1999).
In a recent study, fludeoxyglycose (FDG) uptake after 1 mins of visual stimulation was higher than after 15 mins of continuous stimulation, in which it was attenuated by 28%. This suggests a shift from glycolytic to oxidative glucose metabolism with continued activation (Vlassenko et al, 2006). This may be considered as compatible with the observation that only part of the excess glucose uptake can be accounted for by a release from the brain to the blood of lactate (discussed above, Madsen et al, 1999). Thus, the excess glucose uptake cannot accumulate forever in the brain and a new balance must be achieved. In this context, we have to recall that what one measures is in most instances a deviation of a balance between metabolism and delivery of substances triggered by metabolic changes, and such a deviation may have a time profile by its own. Mintun et al (2004) and Vlassenko et al (2006) hypothesized that, in activated brain, blood flow is modulated by changes in the cytosolicfree nicotinamide adenine dinucleotide (NADH)/NAD+ ratio, related to increased glycolysis —a theory in accord with our hypothesis of the flow increase being driven by an excess demand for glucose.
The Activation-Induced Cerebral Blood Flow Response During Hypoglycemia
If the functionally most important consequence of the rCBF response to activation is an increased glucose supply, hypoglycemia could be expected to augment the CBF response. Using PET and 15O2, Powers et al (1996) found similar increases in the CBF in response to activation (instead of augmentation), when comparing subjects exposed to acute moderate hypoglycemia with normoglycemic subjects. On the basis of their findings, the authors concluded that the increase in rCBF during physiologic brain activation was not regulated by a mechanism that matches local cerebral glucose supply to local cerebral glucose demand. However, there are some inherent limitations with the study by Powers et al (1996). First, no measurements of CMRglc were obtained. This is a key piece of information for judging whether coupling may be present or lacking. There is evidence from human subjects that CMRglc begins to decline at blood glucose levels of ∼3 mmol/L (Lubow et al, 2006). Second, no confirmation was obtained that the stimulus (fingertip vibration) elicited identical electrical responses (equal magnitude neural activations) in the cortex in normoglycemic versus hypoglycemic states. This has some relevance, as even moderate hypoglycemia may alter cerebral electrophysiologic functions (Rosenthal et al, 2001). In fact, Powers et al (1996) reported significant neurobehavioral changes in a group of hypoglycemic subjects (plasma glucose=2.8 mmol/L). This could have affected cortical reactions to somatosensory stimuli, including CMRglc responses. Despite the caveats, these findings seem to imply that the generally close coupling between CBF and glucose utilization may begin to fail when glucose supply becomes limiting and interferes with the capacity to increase the cerebral glucose consumption rate, under conditions of neural activation. Further support for hypoglycemia (plasma glucose=2.2 mmol/L) interfering with sensory processing and brain activity/CBF relationships in human subjects was provided in a recent report by Bie-Olsen et al (2009). Here, the authors concluded that hypoglycemia induces changes in sensory processing in a cognition-independent manner, whereas activation of areas of higher order functions is influenced by cognitive load as well as hypoglycemia.
Recent fMRI studies have indicated that functional activation during hypoglycemia does not augment, but rather attenuates, the BOLD response (Rosenthal et al, 2001; Kennan et al, 2005; Anderson et al, 2006). In the study by Kennan et al (2005), baseline CBF increased 14%, when going from euglycemia to moderate hypoglycemia (plasma glucose= 3.6 mmol/L). It has been earlier shown that increases in baseline CBF can decrease the BOLD signal through limiting the CBF response during subsequent activations (Vazquez et al, 2006; Cohen et al, 2002). This might, in part, explain the lack of neural activation-induced CBF augmentation during hypoglycemia. Nevertheless, while the results of Kennan
In addition, there are other conditions associated with CMRglc/CBF uncoupling. Thus, unlike the hypoglycemia findings, seizure-prone mice displayed increased rCMRglc, in the absence of rCBF changes (Hosokawa et al, 1997), whereas in rats subjected to cortical impact trauma, rCMRglc/rCBF ratios were substantially elevated (Chen et al, 2004).
In conclusion, the studies of hypoglycemia do not support the concept of glucose lack, as the triggering mechanism for the flow increase occurring during functional activation. Despite these and additional findings, evidence of an apparent uncoupling of CBF/CMRglc under mild (hypoglycemia) to severe (seizure; neurotrauma) perturbations does not negate the presence of close coupling under normal physiologic circumstances. Such findings may only be taken as indicating that there are constraints on that linkage.
The Significance of Capillary Heterogeneity for Glucose Transport
For glucose, an increase in CBF in a homogenous vascular bed is an inefficient way to increase its transport across the BBB. Under normal physiologic conditions, the arterio-venous difference for glucose is about 10% and the unidirectional flux close to 20%. That is, approximately half of the glucose entering the brain from the capillaries diffuses back to the blood (Knudsen et al, 1990). With an arterio-venous difference of 10%, the average capillary concentration of glucose would be close to 95% of the arterial concentration. Thus, even an infinitely high increase in CBF, with preserved heterogeneity, would increase the average capillary concentration and thereby the glucose transfer from blood to brain by only 5%. Functional recruitment, in contrast, would be a much more efficient way to increase glucose supply. This is because in capillaries with a low flow, the average concentration of nutrients will approach the tissue level, and net transport will be minimal. Thus, if half of the capillaries had a high and the other half a low-linear velocity, then recruitment with homogenization of CBF to the high value would double the potential for glucose transfer from the blood to the brain. When discussing glucose transfer across the BBB, back flux from the tissue to the blood has also to be taken into consideration. Thus, a drop in peri-capillary glucose concentration might increase the net glucose uptake. Such a drop has been observed in activation studies using magnetic resonance spectroscopy (Chen et al, 1993; Frahm et al, 1996). However, in studies using direct measurements of brain tissue glucose and lactate, an essentially constant glucose concentration was found during activation, whereas lactate increased (Madsen et al, 1999). Thus, it seems that a CBF increase with capillary recruitment could be a key factor responsible for an increased glucose uptake during activation, although models postulating translocation of the endothelial glucose transporter (GLUT1) from intracellular sites to the plasma membrane need to be considered (Cornford et al, 2000).
Oxygen Metabolism—Cerebral Blood Flow Uncoupling
During functional activation studies, using positron emission tomography and the Kety–Schmidt technique did not reveal any major increases in CMRO2 (Fox et al, 1988; Madsen et al, 1995). In contrast, studies using the magnetic resonance BOLD methods has revealed a CMRO2 increase of about a third of the increase in CBF (Davis et al, 1998; Kim et al, 1999). Animal studies have shown that oxygen consumption not only increases during activation, but also decreases during deactivation (Hyder et al, 2001).
It has been hypothesized that this more modest increase in oxygen consumption still can explain the uncoupling of CBF and oxidative metabolism during functional activation because of diffusion limitations for oxygen diffusion limiting oxygen availability in the tissue (Buxton and Frank, 1997; Kuwabara et al, 1992), either because of limited diffusion across the BBB or diffusion limitations within the brain tissue. Below, we will look at these aspects separately and add new evidence from recent studies to classic models of oxygen transport and diffusion (Uludağ et al, 2004; Zheng et al, 2002; Kuschinsky and Paulson, 1992).
Oxygen Limitation Concept
Buxton and Frank (1997) formulated a model, which assumed a low tissue oxygen concentration and no capillary recruitment. This model showed a nonlinear relationship between CBF and oxygen extraction, and, as a consequence, O2 extraction increased to a proportionately smaller degree than CBF did. Similar conclusions were reached by Vafaee and Gjedde (2000). In addition, in a more complex model with non-negligible tissue oxygen tension, thus allowing for back flux from tissue to blood by Valabrègue et al (2003), a non-linear relationship was found. Although these models have attracted much attention, they may still be challenged. In addition, in the following discussion, we will focus on two limitations, which may represent an oversimplification: the assumption of lack of capillary heterogeneity and of lack or limitation of back flux of oxygen from the tissue to the blood.
Oxygen Diffusion Across the Blood–Brain Barrier
To what extent capillary recruitment is capable of increasing tissue oxygen supply is not as simple to quantify as for glucose. An increased effective capillary surface area as well as an increase in oxygen delivery through an appropriate CBF increase would facilitate oxygen transfer across the BBB. Further, functional capillary recruitment will lead to an increased number of transport efficient capillaries in the tissue and thereby to shorter diffusion distances from the capillaries to the more remote part of the tissue. The latter might be of major significance and account for the increased cortical oxygen tension observed during functional activation (Cooper et al, 1966). Only a few studies have measured oxygen transport in the brain. Using the double indicator method, Grieb et al (1983) found no significant diffusion limitation for oxygen from blood to brain tissue. In a similar setup, however, Kassissia et al (1995) could not confirm the observation of Grieb
Thus, the physiologic validity of the model used in the study of Kassissia et al (1995) may be questioned. Their calculation of a capillary permeability surface area product for oxygen of 70 mL/g/min may, therefore, represent a gross underestimation, and oxygen is highly diffusible and rapidly released from hemoglobin. In conclusion, diffusion across the BBB seems not in any likelihood to explain the uncoupling of oxygen metabolism and CBF during activation.
Oxygen Diffusion in Brain Tissue
The oxygen limitation concept states that because of the diffusion gradient in the brain tissue, an increased oxygen metabolism associated with functional activation would, in the absence of a sufficient increase in rCBF, lead to an insufficient oxygen tension in the least oxygenated parts of the cerebral tissue, that is the mitochondria. The oxygen limitation hypothesis has been addressed through sophisticated modeling of cerebral oxygen consumption and delivery (Buxton and Frank, 1997; Zheng et al, 2002). In both models, recruitment or capillary heterogeneity as well as back diffusion of oxygen were not, however, taken into account. Accordingly, Hyder et al (1998) have used another model for oxygen delivery to the brain, in which they include a variable for increased diffusibility of oxygen with increased CBF. With this model, oxygen delivery to the brain is not critical during functional activation.
The old observation of an increased tissue oxygen tension in activated regions (Cooper et al, 1966) would seem non-compatible with the theory of deficient oxygen supply in functional activation. However, the models referred to imply a steeper gradient of the oxygen tension in the tissue, and this may lead to decreased oxygen tension in the cerebral tissue most distant from the capillaries concomitant with an increase of the average tissue oxygen tension. Recent studies have shed some light on the oxygen limitation hypothesis. In hypoxia, both acute and after adaptation to high altitude, the relative rCBF increase during functional activation remains unaltered (Mintun et al, 2001; Law et al, 2002; Tuunanen and Kauppinen, 2006), even though the BOLD response is diminished (Rostrup et al, 2005; Tuunanen and Kauppinen, 2006; Ho et al, 2008). Further, the same degree of deoxygenation of hemoglobin takes place during the passage of the blood through the cerebrovascular bed at high altitude and at sea level, reflecting a lower venous oxygen saturation and tension in view of the lower arterial oxygen saturation at high altitude (Møller et al, 2002).
Oxygenation at the Cellular Level
Detailed studies with high time resolution of the tissue oxygenation tension in rats has revealed an initial dip in oxygen tension during activation, lasting about 2 secs. This was followed by a marked increase of the oxygen tension with a delay of 1–2 secs as compared with the observed flow increase (Ances et al, 2001). Using visual stimulation in cats, and simultaneous recording of oxygen tension and single cell neural spike rate for 4 secs, revealed a decrease in oxygen tension accompanying the neural activity (Thompson et al, 2003). This could correspond to the initial dip in oxygen tension observed in the former study, but might also reflect a longer lasting decrease in oxygen tension in some highly active cells. In contrast to these studies, a recent preliminary report showed that the mitochondrial oxygen tension did not decrease during functional activation, but rather increased (Springett et al, 2007). These studies may give rise to some considerations regarding the spatial-temporal spread function. At the subcellular level, for example mitochondria (in which oxidative metabolism actually takes place), one could envisage that oxygen tension and the concentration of glucose and/or lactate was lower than in the surrounding cellular elements. In such subcellular fractions, oxygen tension might be low and decrease further during activation. However, this would also be the case for glucose and lactate. In fact, diffusion across cellular or subcellular lipid membranes is at least 10-fold larger for oxygen than for glucose and lactate. Thus, a very low oxygen tension in these subcellular compartments might very well be accompanied by similarly low concentrations of glucose and lactate. Considering the temporal relationship between metabolism and flow, a delay in the flow response might be anticipated. Thus, even with an instantaneous relaxation of the tone of the resistance vessels, there will be a delay before vasodilation occurs and new blood has flowed into the capillary bed. With an average capillary transit time of 1–2 secs, one would expect such a delay to be a few seconds in magnitude.
Kasischke
rCBF and rCMRglc coupling: the astrocyte–arteriole connection
Signaling Factors
When attempting to understand the close coupling between rCBF and CMRglc, especially under conditions of increased neural activity, one cannot ignore the astrocyte. One reason is that astrocytes are major glucose-consuming cells. That is, in astrocytes, glycolytic conversion of glucose to lactate seems to be the preferred energy-generating pathway, as opposed to neurons, in which lactate and glucose oxidation may be more predominant (Berkich et al, 2007). Another consideration relates to the well-documented physical association between astrocytic processes (especially endfeet) and cerebral arterioles. Astrocytic endfeet extensively ensheath cerebral arterioles (Straub and Nelson, 2007). Astrocytes physically link neurons and their synapses with the vasculature, and are in a strategic position to convey neuronal signals to the blood vessels. Increased synaptic activity may lead to the release of factors (e.g., glutamate, ATP) that are ‘detected’ by adjacent astrocytes (Figure 2). This initiates a signal that can be transmitted, even over multiple astrocytes, to perivascular glial endfeet, and elicits changes in arteriolar tone. During neural activation states, such astrocytic communication may help to ensure an adequate increase in substrate delivery through signaling both local and upstream arteriolar segments to dilate (Iadecola and Nedergaard, 2007; Xu et al, 2008). A key participant in the link between neural activation, inter-astrocytic communication, increased astrocytic glucose utilization, and vasodilation is Ca2+ (Takano et al, 2006; Scemes and Giaume, 2006; Wang et al, 2006). Ca2+ contribution is thought to arise from ATP and/or glutamate-mediated activation of astrocytic metabotropic purinergic and glutamatergic receptors (P2Y and mGluR, respectively) (Zonta et al, 2003; Nedergaard et al, 2003; Shi et al, 2008).
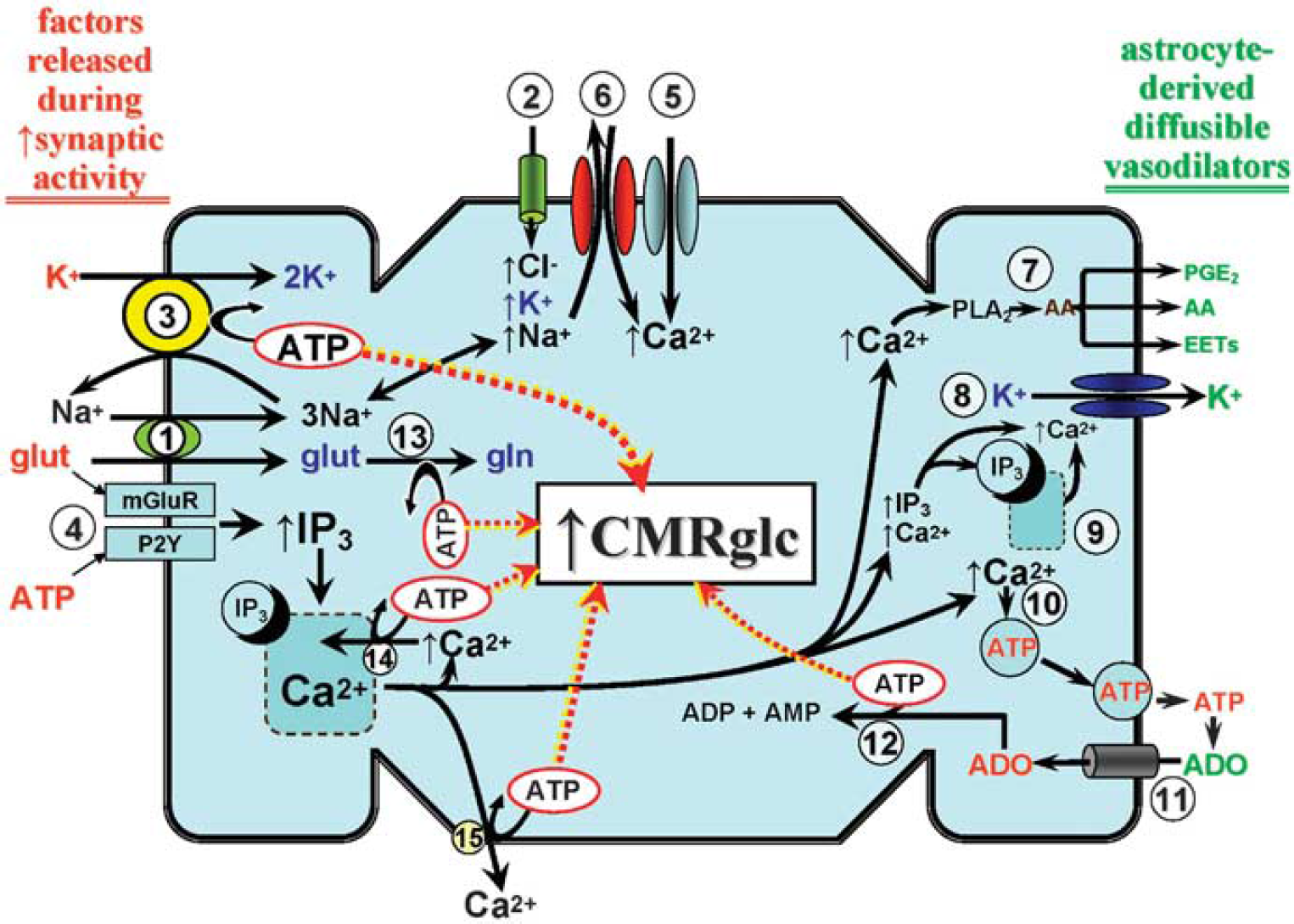
A generalized depiction of the energy cost of neurovascular coupling and its impact on
As illustrated in Figure 2, activation of mGluR or P2Y receptors promotes IP3-evoked release of Ca2+ from intracellular storage sites in the endoplasmic reticulum (ER). This may promote a cascade of events resulting in further increases in intracellular Ca2+ levels through activation of plasma membrane store-operated Ca2+ channels and/or increased activity of the plasma membrane Na+/Ca2+ exchanger operating in the reverse mode (Blaustein et al, 2002; Rojas et al, 2007). Although the increased intracellular [Ca2+] may have an important function both in propagating astrocyte-to-astrocyte signaling, for example through Ca2+-stimulated ATP and glutamate exocytosis (Bezzi et al, 2004; Bowser and Khakh, 2007; Pangrsic et al, 2007) and Ca2+-dependent vasodilation (see Figure 2 and below), this added Ca2+ load is likely to impact on astrocytic energy demand through activation of ATP-dependent Ca2+ extrusion at the plasma membrane and ATP-dependent refilling of ER Ca2+ stores.
Another entity linked to inter-astrocytic communication and regulation of arteriolar tone is K+. Thus, it has been suggested that, during brain activation, a spatial redistribution of K+ may occur, whereby the increases in extracellular [K+] that take place at the sites of increased neuronal activity would be removed by nearby astrocytes, through a variety of influx mechanisms (e.g., inward rectifier K+ [Kir] channels; Na-K-2Cl cotransporters; and Na-K ATPase), and transferred (possibly through multiple astrocytes, through gap junctions) to astrocytic endfeet in which it could be released to the extracellular environment of adjacent cerebral arterioles, through a Ca2+-dependent mechanism primarily involving large-conductance Ca2+-operated K+ (BKCa) channels (Filosa et al, 2006; Straub and Nelson, 2007). Evidence indicates that BKCa channels are particularly concentrated in astrocyte endfoot plasma membranes facing cerebral microvessels (Price et al, 2002). Also expressed in astrocyte endfoot processes are ER IP3 receptors which, when activated, seem to provide the necessary (albeit spatially restricted) Ca2+ increases for BKCa channel opening (Straub et al, 2006). The subsequent interaction of K+, released through endfoot BKCa channels, with Kir channels on arteriolar smooth muscle represents a potentially powerful stimulus for vasodilation (Paulson and Newman, 1987; Filosa et al, 2006).
In addition, as summarized in Figure 2, one must consider the energy cost arising from the increase in extracellular [K+], and its uptake into astrocytes, during periods of enhanced neural activity. The increased extracellular [K+], together with the associated enhanced entry of Na+ through the Na+-K+-2Cl− cotransporter plus the Na/glutamate cotransporter, will stimulate Na-K ATPase activity. This represents a major, if not the principal, demand on glycolytic energy production. It should also be noted that some of the glutamate taken up by the astrocyte will be transferred to neurons in the form of glutamine. The intra-astrocytic conversion of glutamate to glutamine consumes ATP. Finally, the ATP released in the process of inter-astrocytic communication is lost as an energy source. That is, it cannot return to the cell as ATP, as it is rapidly hydrolyzed by ecto-nucleotidases, which are well expressed on astrocytes (Wink et al, 2006; Zimmermann, 2006), ultimately forming adenosine. To limit depletion of intracellular adenine nucleotide levels, adenosine re-enters the astrocyte [through a cell membrane nucleoside transport system (Peng et al, 2005)] to enter adenine nucleotide salvage pathways. The key enzyme of the salvage pathway is adenosine kinase, whose expression is largely confined to astrocytes (Boison, 2008). The energy cost of salvage arises from the ATP consumed in the adenosine kinase-mediated formation of ADP and AMP.
In a compelling study using primary cultures of murine cortical astrocytes, Bernardinelli et al (2004) examined the implications of the appearance of an astrocytic Ca2+ wave, as it impacts on glucose utilization and, by extension, energy production. These authors reported that an electrically evoked induction of a Ca2+ wave was accompanied both by a parallel Na+ wave and a wave of increased glucose uptake as well. Furthermore, the Na+ wave was associated with an increased glutamate uptake, presumably through the Na/glutamate cotransporter. Thus, not only may propagated inter-astrocytic cationic waves occur in response to neural activation, but also parallel ‘waves’ of increased glucose metabolism may occur as well.
Besides its postulated function in BKCa channel-mediated vasodilations, the arrival of the Ca2+ wave at astrocytic endfoot points of contact with arterioles can elicit smooth muscle relaxation through other mechanisms. This includes stimulation of astrocytic Ca2+-activated phospholipase A2 and the release of arachidonic acid (AA). A function for astrocyte-derived AA metabolites, and AA itself, as paracrine factors in neural activation-evoked cerebral vasodilation has been indicated in studies showing neurovascular coupling to be attenuated in the presence of phospholipase A2, cyclooxygenase-1, and/or epoxygenase inhibition (Takano et al, 2006; Wang et al, 2006; Shi et al, 2008). Finally, one cannot ignore the potential vasodilating actions associated with the adenosine that is formed from extracellular ATP. That adenosine may influence brain arterioles indirectly by virtue of interactions with astrocytic adenosine receptors (A2B), perhaps contributing to inter-astrocytic (Ca2+) signaling (Shi et al, 2008) or can act as a paracrine factor, directly influencing vascular smooth muscle adenosine receptors (e.g., A2A, see Meno et al, 2001), leading to vasodilation.
Metabolic Consequences of Signaling
Taken together, the signaling factors given above can place substantial demands on astrocytic energy sources and probably provide a potent stimulus for increasing CMRglc during neural activation. There is strong evidence that the most-used procedures for measuring CMRglc
Signaling and Metabolism: Summary
There is abundant evidence that astrocytes have a central position in neurovascular coupling (Figure 2). Increases in synaptic activity are accompanied by an elevated presence of various signaling entities in the extracellular fluid bathing adjacent astrocytes. These include ATP, K+, and glutamate. ATP and glutamate can interact with receptors on astrocytes, which may give rise to a wave of increased Ca2+ across multiple astrocytes that eventually reaches astrocytic endfeet in contact with cerebral arterioles. The increased extracellular [K+] can enter astrocytes through several routes, in which it may be spatially redistributed and released from endfeet through a Ca2+-dependent process involving BKCa channels, subsequently eliciting arteriolar smooth muscle relaxation mediated by Kir channels. In addition, the extracellular ATP can be rapidly converted to adenosine, another potent signaling molecule and vasodilator. The astrocytic uptake of K+ and glutamate is associated with a rise in intracellular [Na+]. Taken together, the above processes, many of which can be linked directly to arteriolar dilation, place a substantial demand on astrocytic energy sources. In astrocytes, a significant portion of that energy may be derived from non-oxidative glucose consumption. Moreover, the model proposed in Figure 2 could be consistent with a later activation of glycolysis in astrocytes, after neuronal activation, as proposed by Kasischke et al (2004). That is, the astrocyte ATP utilization and subsequent energy replenishment through increased glycolysis might be expected to occur in the immediate post-stimulus recovery phase. Although this may contribute to the closer association between increased CBF and increased CMRglc (as opposed to CMRO2) observed during increased brain activity, it is probably not entirely accurate, as the Kasischke
Concluding remarks
In the above consideration of substrate delivery to the brain during functional activation, we endeavored to take all relevant physiologic players into consideration: diffusibility of oxygen and glucose; lack of classic capillary recruitment, but presence of functional recruitment as a consequence of reduced capillary heterogeneity with increased CBF; as well as the very tight coupling between increases in CBF and CMRglc in contrast to a lower relative increase in oxygen consumption. Further, recent studies of oxidative and non-oxidative glucose metabolism during activation as well as studies on the astrocytic regulation of rCBF response support the view that the disproportional rCBF increase during functional activation relates to the need for sufficient glucose supply. Consequently, the increased oxygen content in the venous blood from the brain observed in the BOLD response is a secondary event to the CBF-glucose coupling. Future research may be directed toward the understanding of the basis for the CBF-glucose coupling—especially why the brain needs an excess glucose uptake during activation.
Footnotes
The authors declare no conflict of interest.