
Abstract
Cytosolic phospholipase A2 (cPLA2) is a key enzyme that mediates arachidonic acid metabolism, which causes cerebral ischemia-induced oxidative injury, blood—brain barrier (BBB) dysfunction, and edema. Recent reports have shown that p38 mitogen—activated protein kinase (MAPK) is related to phosphorylation and activation of cPLA2 and release of arachidonic acid. However, involvement of the p38 MAPK pathway in cPLA2 activation and of reactive oxygen species in expression of p38 MAPK/cPLA2 after ischemia—reperfusion injury in the brain remains unclear. To address these issues, we used a model of transient focal cerebral ischemia (tFCI) in rats. Western blot analysis showed a significant increase in expression of phospho-p38 MAPK and phospho-cPLA2 in rat brain cortex after tFCI. Activity assays showed that both p38 MAPK and cPLA2 activation markedly increased 1 day after reperfusion. Intraventricular administration of SB203580 significantly suppressed activation and phosphorylation of cPLA2 and attenuated BBB extravasation and subsequent edema. Moreover, overexpression of copper/zinc-superoxide dismutase remarkably diminished activation and phosphorylation of both p38 MAPK and cPLA2 after reperfusion. These findings suggest that the p38 MAPK/cPLA2 pathway may promote BBB disruption with secondary vasogenic edema and that superoxide anions can stimulate this pathway after ischemia—reperfusion injury.
Keywords
Introduction
Blood—brain barrier (BBB) disruption is a critical event during cerebral ischemia, and is followed by an influx of water, leading to vasogenic edema and secondary brain injury. Cerebral edema is a major risk for life-threatening complications after acute ischemic stroke, but treatment is limited. As such, the BBB is one of the major targets for understanding mechanisms that mediate cerebral ischemic damage.
Arachidonic acid (AA) causes BBB dysfunction and subsequent brain edema (Chan and Fishman, 1978; Papadopoulos et al, 1989; Unterberg et al, 1987). Arachidonic acid can also be converted to inflammatory lipid mediators, eicosanoids, through the lipoxygenase or cyclooxygenase (COX) pathway (Smith et al, 1996), which play a significant role in brain edema. Several studies have shown that inhibition of AA metabolism attenuates cerebral ischemia-induced oxidative injury, BBB dysfunction, edema, and infarction (Adibhatla and Hatcher, 2003; Rao et al, 1999; Watanabe and Egawa, 1994). Cytosolic phospholipase A2 (cPLA2) mediates AA metabolism and is related to the production of eicosanoids, such as prostaglandins and leukotrienes. Cytosolic phospholipase A2 is regulated by intracellular calcium and translocates from the cytosol to the membrane (Hirabayashi et al, 1999). The knockout of cPLA2 has been reported to reduce infarction and edema and to improve neurologic scores after ischemic brain injury in mice (Bonventre et al, 1997; Tabuchi et al, 2003). A PLA2 inhibitor attenuated ischemia-like injury in rat hippocampus (Arai et al, 2001). Therefore, the cPLA2-induced AA cascade is harmful to the brain. Activation of cPLA2 is related to several mechanisms, including activation of mitogen-activated protein kinase (MAPK) and protein kinase C (Lin et al, 1993; Xu et al, 2002). Several reports have shown that p38 MAPK is linked to activation and phosphorylation of cPLA2 and AA release (Coulon et al, 2003; Waterman et al, 1996). However, involvement of the p38 MAPK pathway in cPLA2 activation after ischemia—reperfusion injury in the brain remains unclear.
Reactive oxygen species, especially superoxide anions, are key components in ischemia—reperfusion injury and can cause direct oxidative damage and contribute to BBB disruption and development of brain edema. Our previous studies have shown that copper/zinc-superoxide dismutase (SOD1)−/−mice are highly susceptible to focal cerebral ischemic reperfusion, with exacerbated vasogenic edema, and have a higher mortality rate than their wild-type (WT) littermates (Gasche et al, 2001; Kondo et al, 1997). Although it has been reported that oxidative ischemic injury is involved in cPLA2 enzymatic activation or immunoreactivity (Bonventre et al, 1997; Saluja et al, 1997; Stephenson et al, 1999), whether SOD1 affects the p38 MAPK/cPLA2 pathway in BBB dysfunction after transient focal cerebral ischemia (tFCI) remains unclear. The aim of this study was to clarify the deleterious role of the p38 MAPK/cPLA2 pathway in BBB permeability and edema formation in the brain after tFCI and the relationship between the p38 MAPK/cPLA2 signaling pathway and SOD1.
Materials and methods
Focal Cerebral Ischemia
All experimental procedures involving animals were approved by the Administrative Panel on Laboratory Animal Care of Stanford University and were conducted in accordance with the recommendations provided in the NIH Guide for the Care and Use of Laboratory Animals. During surgical preparation, male Sprague—Dawley rats (250 to 300 g) were anesthetized with 2% isoflurane in a mixture of 70% N2O and 30% O2 under spontaneous breathing. The left external carotid artery was exposed through a midline cervical incision and its branches were electrocoagulated. A 22-mm 3-0 surgical monofilament nylon suture, blunted at the end, was introduced into the left internal carotid artery through the external carotid artery stump, according to our previous report (Okuno et al, 2004). Body temperature was maintained at 37°C, using a heating pad, during the surgical procedure for middle cerebral artery occlusion (MCAO). After 90 mins of MCAO, cerebral blood flow was restored by removal of the nylon thread. Blood samples were collected from the tail artery before and during MCAO and just after reperfusion for measurement of pH, pO2, and pCO2. Sham control animals were treated in the same way without MCAO.
SOD1 Transgenic Rats
Heterozygous SOD1 transgenic rats with a Sprague—Dawley background, carrying human SOD1 genes with a four- to six-fold increase in copper/zinc-SOD, were derived from the founder stock and further bred with WT Sprague—Dawley rats to generate heterozygous rats as previously described by our group (Chan et al, 1998). The phenotype of the SOD1 transgenic rats was identified by isoelectric focusing gel electrophoresis. There were no observable phenotypic differences, including in the cerebral vasculature, between the transgenic rats and WT littermates.
Drug Injection
For intracranial surgery, the rats were anesthetized and placed on a stereotaxic apparatus. The scalp was incised on the midline and the skull was exposed. Thirty minutes before the ischemic insult, a total of 10 μL of 100 μmol/L of SB203580 (100 μmol/L dissolved in 1% dimethyl sulfoxide), a specific p38 inhibitor (Calbiochem, San Diego, CA, USA), or the vehicle (1% dimethyl sulfoxide) were injected stereotaxically into the right lateral cerebroventricle (coordinates with reference to bregma: 1.4 mm lateral, 0.8 mm posterior, 3.6 mm deep) using a 26-G Hamilton microsyringe (80330; Hamilton Company, Reno, NV, USA).
Western Blot Analysis
The cortex of the affected side was quickly removed at 6 h and 1, 3, and 7 days of reperfusion. The tissue was homogenized and sonicated in homogenizing buffer (250 mmol/L sucrose, 20 mmol/L HEPES, pH 7.4 with KOH, 100 mmol/L NaCl, 2 mmol/L EDTA, 1% protease inhibitor cocktail (Sigma-Aldrich, St Louis, MO, USA)). The homogenate was centrifuged (1,000g, 15 mins, 4°C) and the resulting supernatant was used for quantification. Protein concentrations were determined and equal amounts of protein were loaded per lane after adding the same volume of Tris-glycine sodium dodecyl sulfate sample buffer (Invitrogen, Carlsbad, CA, USA). Sodium dodecyl sulfate-polyacrylamide gel electrophoresis was performed on a Tris-glycine gel (Invitrogen) and then transferred to a polyvinylidene difluoride membrane (Invitrogen). The primary antibodies were 1:1,000 dilution of the anti-phospho-p38 (Thr180/Tyr182) antibody, 1:2,000 dilution of the anti-p38 antibody, 1:500 dilution of the anti-phospho-cPLA2 (Ser505) antibody, 1:500 dilution of the anti-cPLA2 antibody (Cell Signaling Technology, Beverly, MA, USA), 1:1,000 dilution of the anti-COX-2 antibody (Cayman Chemical, Ann Arbor, MI, USA), and 1:10,000 dilution of β-actin (Sigma-Aldrich). Western blots were performed with horseradish peroxidase-conjugated anti-rabbit immunoglobulin G or anti-mouse immunoglobulin G (Cell Signaling Technology) using enhanced chemiluminescence western blotting detection reagents (Amersham International, Buckinghamshire, UK). The film was scanned with a GS-700 imaging densitometer (Bio-Rad Laboratories, Hercules, CA, USA) and the results were quantified using Multi-Analyst software (Bio-Rad).
p38 Kinase Assay
p38 activity was measured with immune complex protein kinase assays, according to the manufacturer's protocol (Cell Signaling Technology). Samples were prepared as described in the section Western blot analysis. Equal amounts of protein prepared from each tissue homogenate were incubated overnight at 4°C with immobilized phosphorylated p38 MAPK monoclonal antibody. After centrifugation, the pellets were suspended using kinase buffer (25 mmol/L Tris (pH 7.5), 5 mmol/L glycerol phosphate, 2 mmol/L dithiothreitol, 0.1 mmol/L Na3VO4, 10 mmol/L MgCl2), and then glutathione S-transferase-activating transcription factor-2 was used as a substrate for the p38 kinase assay at 30°C for 30 mins. p38 activity was measured by western blot.
cPLA2 and COX-2 Activity
The enzymatic activity of both cPLA2 and COX-2 was measured using specific cPLA2 or COX assay kits (Cayman Chemical) as directed by the manufacturer. Animals were anesthetized and killed at 6 h and 1, 3, and 7 days of reperfusion by transcardial perfusion with a phosphate-buffered saline solution, pH 7.4, containing heparin. Each ipsilateral brain cortex was isolated and homogenized in a lysis buffer, then chilled on ice for 30 mins and centrifuged at 10,000g for 15 mins at 4°C. Cytosolic phospholipase A2 activity was measured using arachidonyl Thio-PC as a substrate. To avoid the measurement of secretory PLA2 and Ca2+-independent PLA2, the secretory PLA2-specific inhibitor thioetheramide-PC and the Ca2+-independent PLA2-specific inhibitor bromoenol lactone were added to the samples before the assay. Activity was calculated by measuring the absorbance at 414 nm, using the 5,5'-dithio-bis(2-nitrobenzoic acid) extinction coefficient of 10.66 per mmol/L per cm, and reported as nmol/min per gram of protein. The peroxidase activity of COX was assayed colorimetrically by monitoring the appearance of oxidized N,N,N′,N′-tetramethyl-p-phenylenediamine at 590 nm. A COX-1-specific inhibitor, SC-560, was used to distinguish COX-2 activity from COX-1 activity.
Evans Blue Extravasation
One hour before the animals were killed, 4 mL/kg of 2% Evans blue (Sigma-Aldrich) in normal saline was injected into every animal. The animals were anesthetized and perfused with heparinized saline. For quantitative measurement of Evans blue leakage, the ipsilateral hemispheric tissue was removed and homogenized in 3 mL of N,N-dimethylformamide (Sigma-Aldrich), then incubated for 20 h at 55°C, and centrifuged at 20,000g for 20 mins (Kim et al, 2003). The level of Evans blue was quantitatively determined in the supernatants at 620 nm using a spectrophotometer (Molecular Devices, Sunnyvale, CA, USA).
2,3,5-Triphenyltetrazolium Chloride Staining
The animals were decapitated at 3 days and the brain slices were stained by immersion in 2% 2,3,5-triphenyltetrazolium chloride at 37°C and fixed in 10% buffered formalin overnight. The area of ischemic lesion for each section was measured using an image analysis system (NIH Image software). Edema volume was determined by subtracting the nonischemic cortical or striatal volume from the ischemic volume (Maier et al, 1998).
Examination of Neurologic Symptoms
The neurologic symptoms of each animal were examined by a blinded observer 3 days after reperfusion using a scoring system based on the detection of hemiplegia and abnormal posture (Bederson et al, 1986). The neurologic deficits were scored as follows: 0, normal; 1, forelimb flexion; 2, decreased resistance to lateral push and forelimb flexion; 3, same behavior as score 2, but with circling.
Statistical Analysis
The data are expressed as mean ± s.d. A one-way analysis of variance followed by the Bonferroni/Dunn post hoc test was used to compare the results of the physiologic data, western blot analysis, activity assay, Evans blue extravasation, and 2,3,5-triphenyltetrazolium staining. Neurologic scores were analyzed by the Mann—Whitney non-parametric test. A P-value < 0.05 was considered statistically significant.
Results
Physiologic Data
There was no significant difference in physiologic parameters such as rectal temperature, mean arterial blood pressure, pH, pCO2, and pO2 among the vehiclend drug-treated groups and the SOD1 transgenic rats before or during MCAO (30 mins after MCAO) or just after reperfusion (data not shown).
Activation and Phosphorylation of p38 After tFCI in WT Rats
To investigate the activation and phosphorylation of p38 after tFCI in WT rats, we performed an activity assay and western blot analysis. p38 activity was measured using activating transcription factor-2 as the kinase substrate. For the western blot analysis, we used a specific antibody against phospho-p38 (Thr180/Tyr182) and p38. p38 activation significantly increased from 6 h to 3 days after reperfusion and peaked at 1 day (Figure 1A, P < 0.01). The protein level of p38 phosphorylation was also significantly increased from 6 h to 3 days after reperfusion. Expression of total p38 was sustained at the same level until 1 day and was significantly increased from 3 to 7 days (Figure 1B).
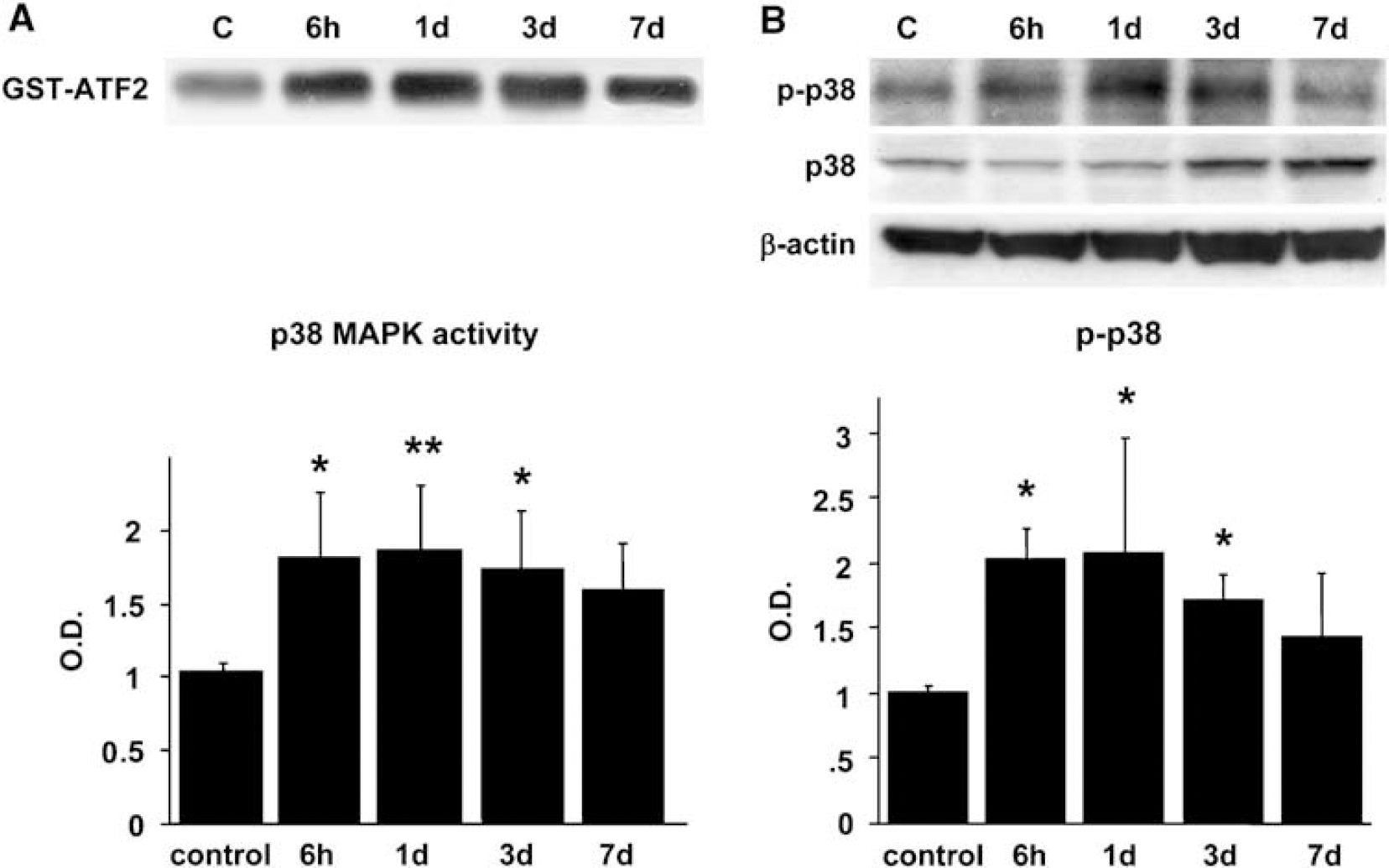
Time course of p38 MAPK activity and expression of phospho-p38 MAPK (p-p38) after reperfusion in WT rats. (
Activation and Expression of cPLA2 and COX-2 After tFCI in WT Rats
We then investigated whether tFCI affects activation and expression of phospho-cPLA2 and COX-2. Cytosolic phospholipase A2 activity was markedly increased 1 day after reperfusion and returned to the basal level at 3 days (Figure 2A, P < 0.01). For protein levels, phospho-cPLA2 (Ser505) was significantly increased at 3 days (Figure 2A, P < 0.01). Total cPLA2 was increased from 3 to 7 days after reperfusion (Figure 2A). Cyclooxygenase-2 activity was significantly increased 1 day after reperfusion (Figure 2B, P < 0.05), whereas its protein level was increased 1 and 3 days after reperfusion (Figure 2B, P < 0.01).
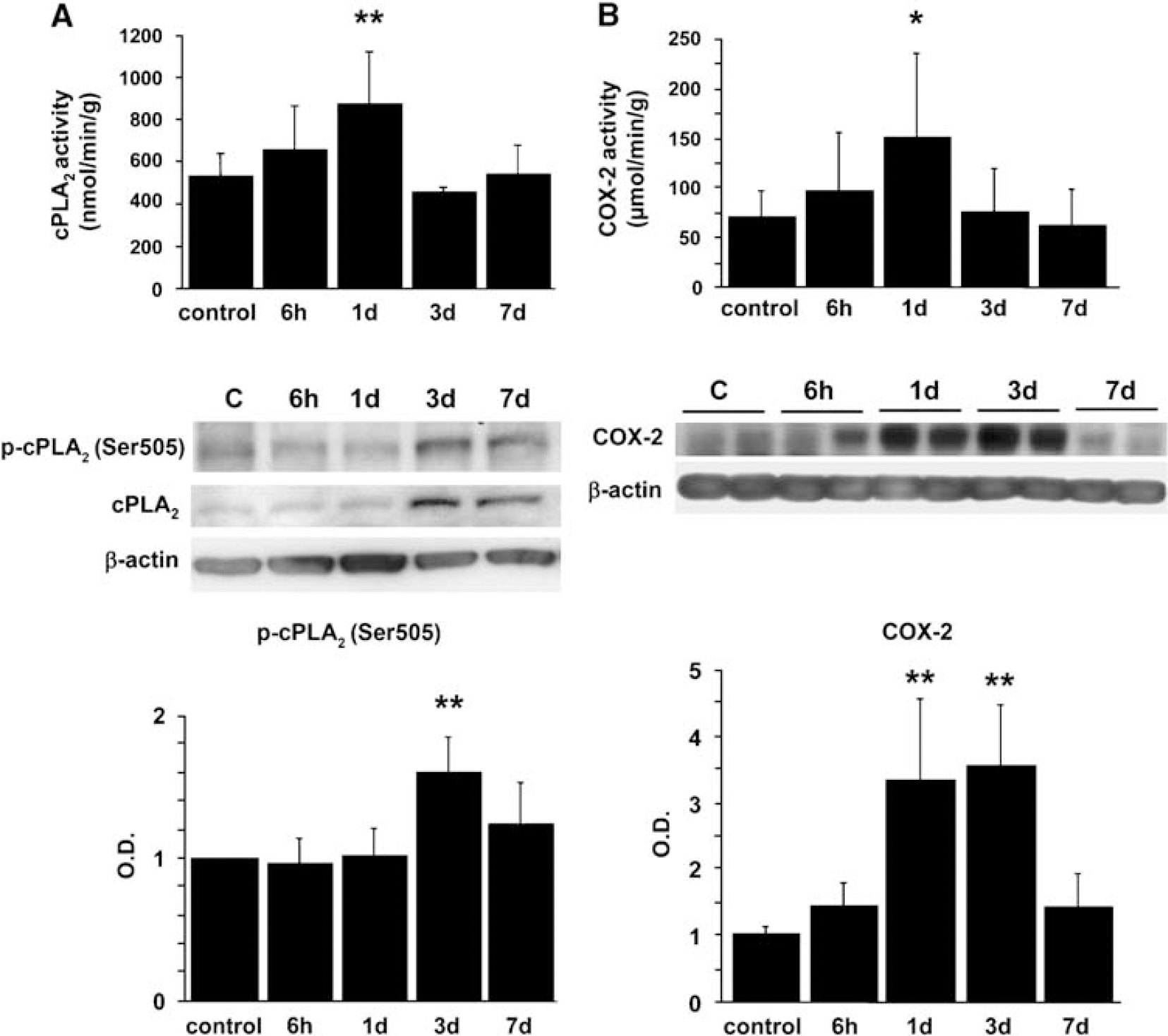
Time course of cPLA2 activity and expression of phospho-cPLA2 (Ser505) (p-cPLA2) after reperfusion in WT rats. (
The p38 Inhibitor SB203580 Blocked cPLA2, Attenuated BBB Permeability and Edema and Infarct Volumes, and Improved Neurologic Function After tFCI
To investigate involvement of the cPLA2 pathway as a downstream effector of the p38 MAPK signaling pathway in the WT rats after tFCI, we performed a study using SB203580, a specific inhibitor of p38. Administration of SB203580 significantly attenuated p38 MAPK activity 1 and 3 days after reperfusion without affecting p38 MAPK phosphorylation (Figure 3A, P < 0.01 and P < 0.05, respectively), because the phosphorylated form of p38 is not inhibited by SB203580 (Larsen et al, 1997). We also examined cPLA2 activation and phospho-cPLA2 (Ser505) expression after administration of SB203580. One day after reperfusion, cPLA2 activation was decreased in the SB203580-treated animals compared with the vehicle-treated animals (Figure 3B, P < 0.01). SB203580 also significantly inhibited phospho-cPLA2 (Ser505) protein levels 3 days after reperfusion (Figure 3B, P < 0.05). Cyclooxygenase-2 activation and expression were also measured. One day after reperfusion, neither the activity nor the protein level of COX-2 was affected by the p38 inhibitor compared with the controls (Figure 3C), whereas SB203580 significantly inhibited the COX-2 protein level at 3 days (Figure 3C, P < 0.01).
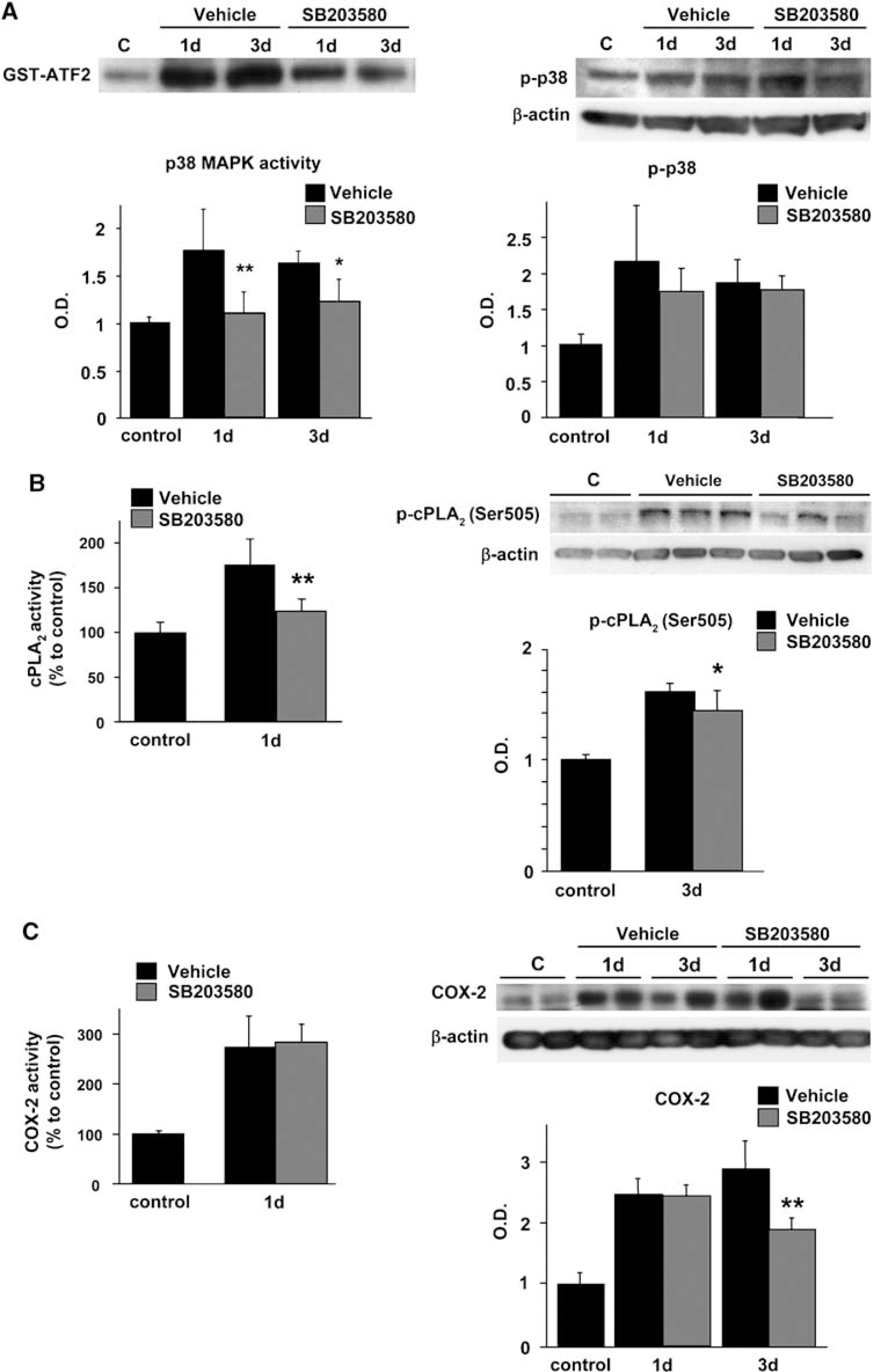
Brain p38 MAPK, cPLA2, and COX-2 after reperfusion and treatment with the vehicle or SB203580 in WT rats. (
We then examined BBB extravasation in the vehicle-treated animals and the SB203580-treated animals. Changes in BBB leakage indicated that BBB breakdown started at 6 h and significantly increased 1 and 3 days after reperfusion (Figure 4A, P < 0.01). Blood—brain barrier leakage was significantly decreased in the SB203580-treated animals compared with the vehicle-treated animals both 1 and 3 days after reperfusion (Figures 4B and 4C, P < 0.01 and P < 0.05, respectively). p38 inhibition significantly attenuated infarct and edema volumes in the cortex 3 days after tFCI (Figures 5A and 5B) and the neurologic deficits improved in the treated rats compared with their untreated counterparts (Figure 5C).
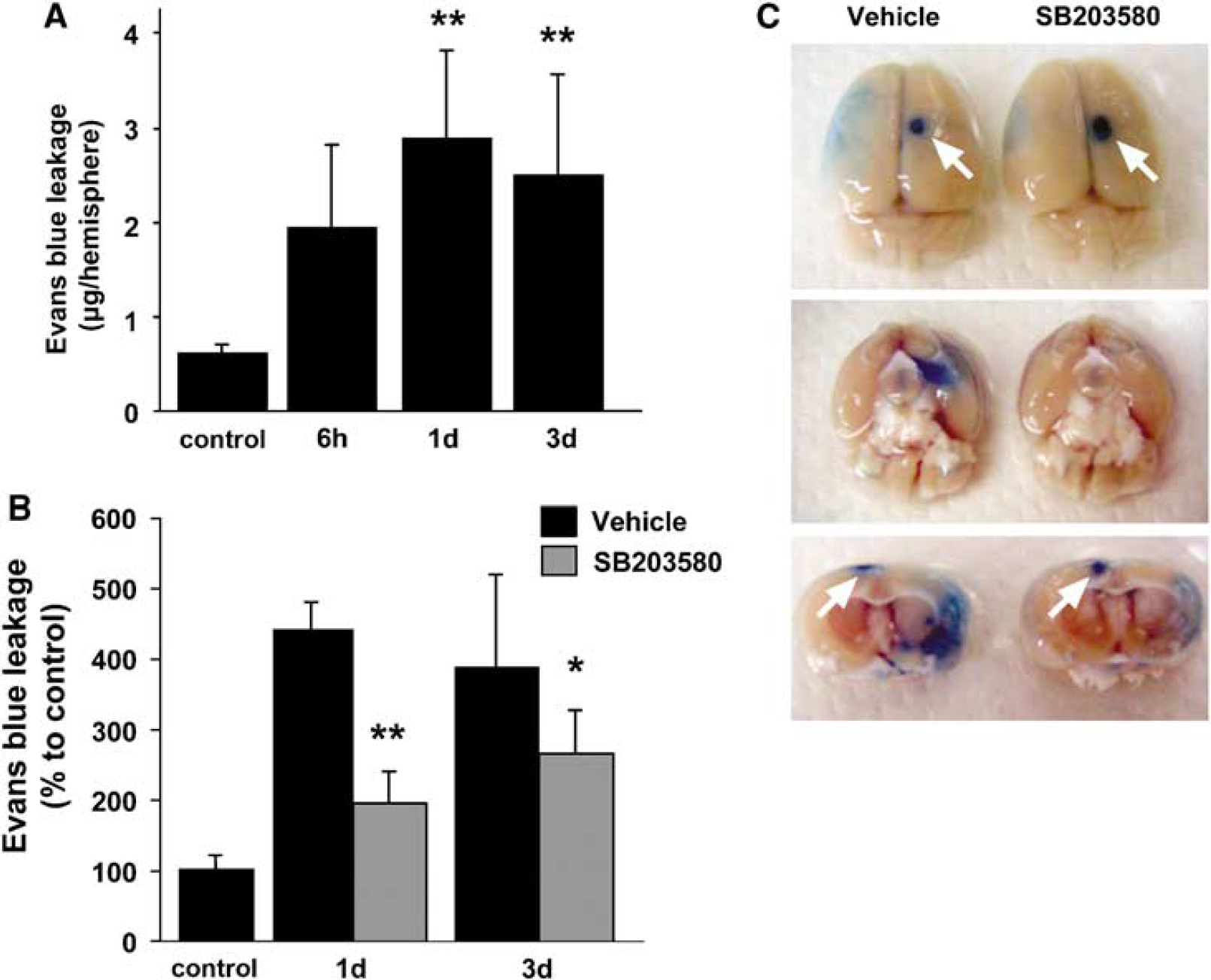
Quantitative analysis of Evans blue leakage in WT rats after tFCI. (
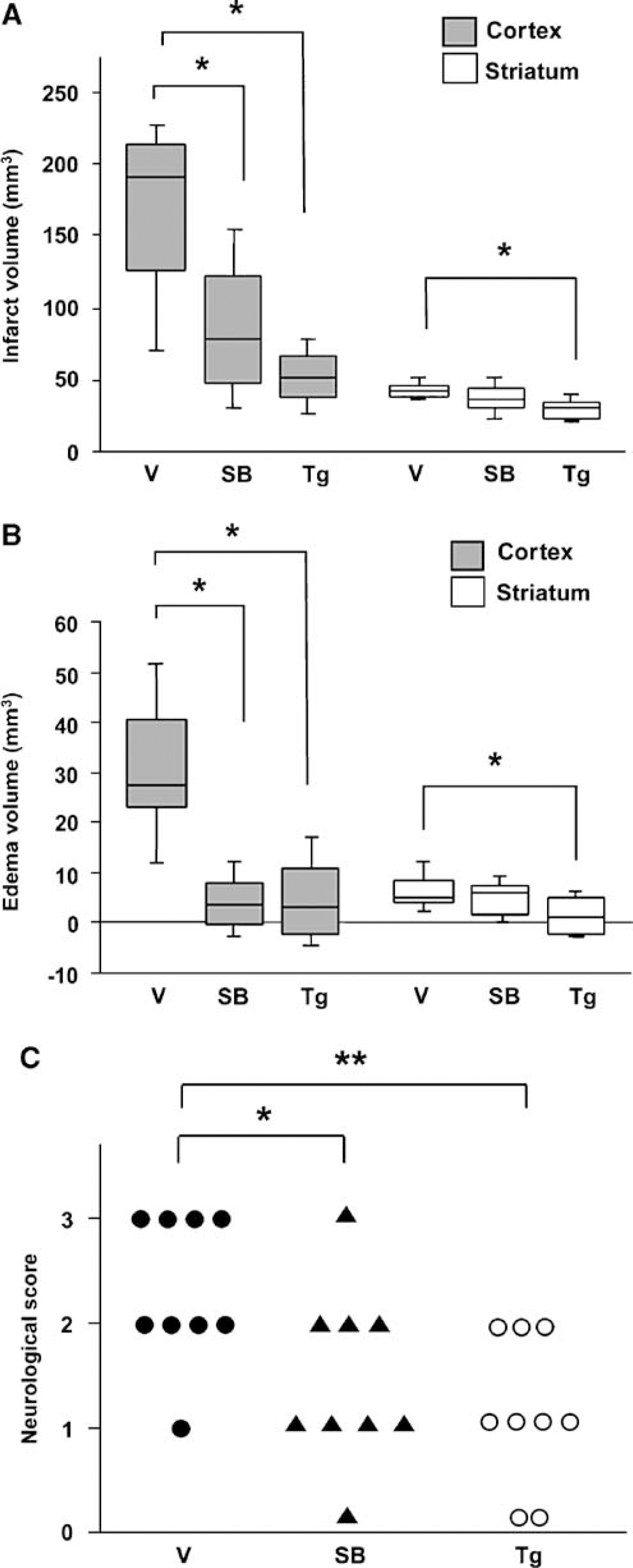
Infarct volume (
Activation and Phosphorylation of p38 and cPLA2 Decreased in the SOD1 Transgenic Rats After tFCI
We have reported on the effects of SOD1 overexpression on cerebral edema formation and BBB permeability after tFCI in mice (Kondo et al, 1997). In the present study, we used SOD1 transgenic rats to investigate whether these effects are mediated by the p38 MAPK/cPLA2 signaling pathway. A kinase assay and western blot analysis showed that p38 activity and phospho-p38 were significantly decreased 1 and 3 days after reperfusion in the SOD1 transgenic rats compared with the WT rats (Figure 6A, P < 0.01 and P < 0.05, respectively). Moreover, activation of cPLA2 and COX-2 was also significantly decreased 1 day after reperfusion (Figure 6B, P < 0.01). For protein levels, SOD1 significantly inhibited phospho-cPLA2 (Ser505) at 3 days (Figure 6C, P < 0.05) and COX-2 expression 1 and 3 days after reperfusion (Figure 6C, P < 0.01 and P < 0.05, respectively).
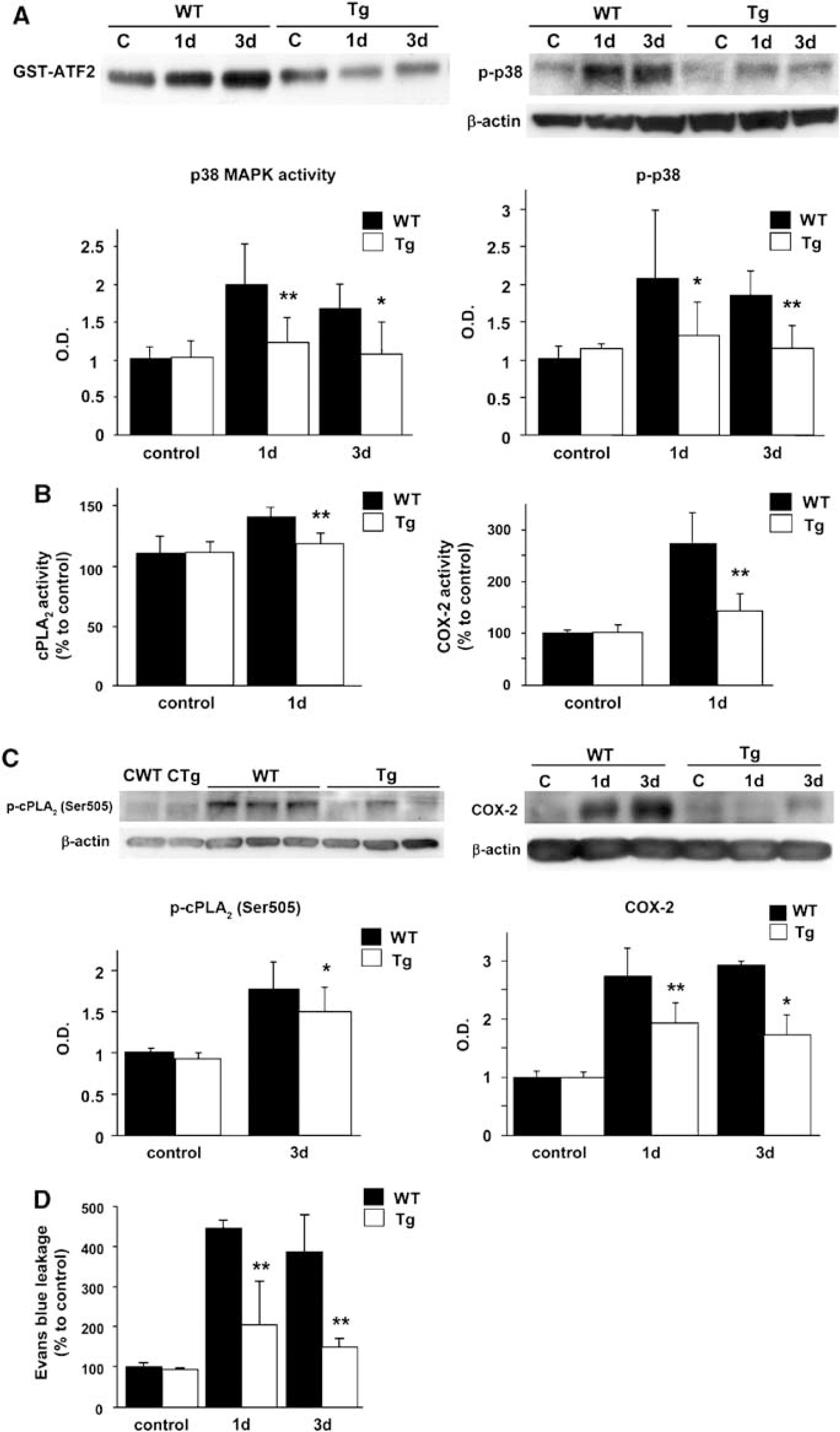
Brain p38 MAPK, cPLA2, and COX-2 after reperfusion in WT and SOD1 transgenic rats. (
SOD1 Attenuated BBB Permeability and Edema and Infarct Volumes, and Improved Neurologic Scores After tFCI
We then examined BBB leakage in the SOD1 transgenic and WT brains 1 and 3 days after reperfusion. Blood—brain barrier leakage was significantly decreased in the transgenic animals compared with the WT animals both 1 and 3 days after reperfusion (Figure 6D). SOD1 significantly decreased infarct and edema volumes not only in the cortex, but also in the striatum 3 days after tFCI (Figures 5A and 5B) and improved neurologic symptoms (Figure 5C).
Discussion
We show that activation and phosphorylation of cPLA2 were prevented by SB203580, which ameliorated BBB breakdown and reduced subsequent brain damage after tFCI. Moreover, there was a remarkable decrease in activation and phosphorylation of both p38 MAPK and cPLA2 after ischemia—reperfusion injury in transgenic rats that overexpress SOD1.
PLA2 exists in the cytosol or close to the membrane of the cell and can be divided into two major classes: Ca2+-independent PLA2 and Ca2+-dependent PLA2, which includes cPLA2 and secretory PLA2. The 85-kDa cPLA2 is a specific producer of AA from the sn-2 position of phospholipids and provides the precursor of the eicosanoids in many types of cells in response to several types of stimulation (Leslie, 1997). Cytosolic phospholipase A2 is regulated by transcription and translation to the membrane, is activated by a low concentration of intracellular Ca2+, and is mediated by MAPKs and protein kinase C (Lin et al, 1993; Xu et al, 2002).
Mitogen-activated protein kinases have crucial roles in signal translocation from the cell surface to the nucleus by regulating cell death and survival. Extracellular signal-regulated kinase is activated in response to several kinds of growth factors, intracellular calcium increase, and glutamate receptor stimulation. In contrast, c-Jun N-terminal kinase and p38 MAPK are activated in response to a variety of cellular stresses, such as DNA damage, heat-shock protein, or inflammatory cytokines, and they modulate expression of several transcription factors. These three kinds of signals are involved in oxidative stress. Although recent studies in animal models have shown that expression of p38 MAPK changes in ischemic brain tissue and that inhibition of the p38 MAPK pathway can improve the outcome of ischemic brain injury by prevention of cell death (Irving and Bamford, 2002; Sugino et al, 2000), the downstream targets of p38 kinase have yet to be identified. Several reports have shown that p38 MAPK is related to activation and phosphorylation of cPLA2 and release of AA in thrombin-activated platelets and collagen-activated platelets (Börsch-Haubold et al, 1997), tumor necrosis factor-stimulated neutrophils (Waterman et al, 1996), and rat cortical neurons (Kriem et al, 2005). However, after ischemia—reperfusion injury in the brain, the role of the p38 MAPK pathway in cPLA2 activation is unclear and there are very few data on the protein level of cPLA2 or on its phosphorylation. To investigate these issues, we used a rat transient MCAO model and examined activation and expression of p38 MAPK and cPLA2. We also investigated COX-2 activation and expression, which are also main steps in the AA cascade and are functionally coupled to cPLA2 (Bosetti and Weerasinghe, 2003). Western blot analysis and activity assays showed that activation and phosphorylation of p38 MAPK increased from 6 h and were sustained until 3 days after reperfusion. These results match those of other studies showing that p38 MAPK has a delayed and long duration of activation and phosphorylation after ischemic insult (Krupinski et al, 2003; Piao et al, 2002). We also found that both cPLA2 and COX-2 activity significantly increased at 24 h and that for the protein levels, an increase in COX-2 expression was observed at 24 h and was maintained at a high level up to 72 h, whereas phospho-cPLA2 (Ser505) significantly increased 72 h after reperfusion. We then determined BBB permeability brain edema, infarction, and neurologic function after tFCI in the WT rats. Blood—brain barrier leakage occurred 6 h after reperfusion and significantly increased at 24 to 72 h. Brain edema and infarct volume were increased at 72 h compared with the controls, and led to deterioration of neurologic function.
We have shown here that the increased enzyme activity or immunoreactivity of p38 MAPK, cPLA2, and COX-2 after tFCI is almost consistent with the occurrence of a BBB breakdown in this disease. Although there was a discrepancy between the time course of cPLA2 activity and phosphorylation, and although cPLA2 phosphorylation at Ser505 was delayed compared with its activation, the reason for these gaps is unclear. Although p38 kinase is involved in phosphorylation of cPLA2 on Ser505 in the cell, cPLA2 is also phosphorylated by p38 kinase on four residues, namely Ser427, Ser454, Ser505, and Ser727 (Hefner et al, 2000). In the ischemic brain, activation of cPLA2 might need phosphorylation, except for Ser505, because the role of its phosphorylation in regulation of cPLA2 is dependent on the cell type and agonist (You et al, 2005). Furthermore, Hefner et al (2000) suggested that phosphorylation of cPLA2 is less important for its activation when intracellular calcium rises to a high and sustained level. Ser505 phosphorylation of cPLA2 might not be required for cPLA2 activation and AA release at an early phase (24 h) but at a late phase (72 h) of BBB disruption after ischemia—reperfusion in the brain.
The intraventricular administration of SB203580 significantly suppressed activation and phosphorylation of cPLA2 and this pharmacological inhibition of p38 MAPK led to a reduction in BBB disruption, edema, and infarct volume. Blockade of the p38 pathway contributed to reduced injury in cerebral tissue, which led to improved neurologic status. Although AA release is converted to prostaglandins by COXs, including COX-2, SB203580 did not inhibit either the COX-2 protein level or its activation at 24 h, which may mean that COX-2 induction is related to other signal transductions, but not through the p38 MAPK pathway in the early phase (24 h) of BBB disruption after reperfusion. These results suggest that activation and phosphorylation of cPLA2 occur in a p38 MAPK-dependent manner and promote damage in the brain cortex after ischemia—reperfusion injury. However, COX-2 expression was inhibited by SB203580 72 h after reperfusion. It has also been reported that p38 mediates COX-2 expression (Fiebich et al, 2000). The significant improvement in BBB breakdown caused by SB203580 at 72 h might be correlated not only to inhibition of phospho-cPLA2 (Ser505), but also to COX-2 expression, which is linked to the suppression of stress kinases and antiinflammatory effects. According to previous in vivo studies, a marked induction of cPLA2 was observed in activated astrocytes and microglia in the hippocampal CA1 subregion 72 h after ischemia (Clemens et al, 1996), and monocytes and macrophages that localized to the core of the infarction were strongly cPLA2-immunoreactive, whereas cPLA2-immunoreactive astrocytes were present in the penumbra 24 h after permanent focal cerebral ischemia (Stephenson et al, 1999). However, those studies examined only the total protein of cPLA2, not its phosphorylation. Our data showed a significant increase in phospho-cPLA2 (Ser505) and total cPLA2 in the cortex 72 h after tFCI by western blotting. This is coincident with the time of activation of astrocytes or microglia, which are implicated in BBB breakdown or inflammation after focal ischemia. Moreover, those cells are also the main types in p38 MAPK signaling. Therefore, we postulate that at a late phase of reperfusion (72 h), astrocytes or microglia are the main cell types in the phosphorylation of cPLA2 on Ser505, which is also implicated in BBB disruption or inflammation after tFCI. Further studies are required to determine the cellular origin and localization of cPLA2 and phospho-cPLA2 at each time point after ischemia—reperfusion injury.
Our previous studies revealed that SOD1 has protective effects against ischemic damage (Chan et al, 1998; Kinouchi et al, 1991; Kondo et al, 1997). Moreover, it is well known that oxidative stress strongly activates PLA2 (Bonventre et al, 1997; Duane et al, 1991; Suzuki et al, 1997). In this study, we also used rats that overexpress SOD1 to focus on the role of the p38/cPLA2 pathway in the neuroprotective effect of SOD1 and investigated whether oxidative stress is imposed on this pathway after ischemia—reperfusion injury. Overexpressed SOD1 significantly decreased activation and expression of both p38 and cPLA2 and alleviated BBB leakage at each time point, suggesting that superoxide anion production after tFCI mediates cPLA2 regulation through p38 MAPK, which has been linked to oxidative stress in various cell types. In addition, although SB203580 inhibited COX-2 expression only at 72 h, overexpression of SOD1 attenuated activation and expression of COX-2 at two time points after reperfusion. Finally, rats with SOD1 overexpression had smaller infarction and less edema in both the cortex and striatum and fewer neurologic deficits. These results support our hypothesis; however, this robust protection compared with SB203580 treatment may suggest that SOD1 could mediate numerous other mechanisms after ischemia. Future in-depth studies using an inhibitor of cPLA2 or genetic approaches are required.
In summary, cPLA2 is regulated in a p38 MAPK-dependent manner in the ischemic brain cortex, and oxidative stress might mediate BBB disruption through the p38 MAPK/cPLA2 pathway after reperfusion following tFCI. This study may provide a new therapeutic strategy for preventing BBB dysfunction induced by oxidative brain injury.
Footnotes
None of the authors has any conflict of interest.