
Abstract
Hemoglobin (Hb) released from extravasated erythrocytes is implicated in brain edema after intracerebral hemorrhage (ICH). Hemoglobin is a major component of blood and a potent mediator of oxidative stress after ICH. Oxidative stress and matrix metalloproteinases (MMPs) are associated with blood–brain barrier (BBB) dysfunction. This study was designed to elucidate whether Hb-induced oxidative stress contributes to MMP-9 activation and BBB dysfunction in vivo. An intracerebral injection of Hb into rat striata induced increased hydroethidine (HEt) signals in parallel with MMP-9 levels. In situ gelatinolytic activity colocalized with oxidized HEt signals in vessel walls, accompanied by immunoglobulin G leakage and a decrease in immunoactivity of endothelial barrier antigen, a marker of endothelial integrity. Administration of a nonselective MMP inhibitor prevented MMP-9 levels and albumin leakage in injured striata. Moreover, reduction in oxidative stress by copper/zinc-superoxide dismutase (SOD1) overexpression reduced oxidative stress, MMP-9 levels, albumin leakage, and subsequent apoptosis compared with wild-type littermates. We speculate that Hb-induced oxidative stress may contribute to early BBB dysfunction and subsequent apoptosis, partly through MMP activation, and that SOD1 overexpression may reduce Hb-induced oxidative stress, BBB dysfunction, and apoptotic cell death.
Keywords
Introduction
Nontraumatic intracerebral hemorrhage (ICH) accounts for 2 million (10% to 15%) of about 15 million strokes worldwide each year. Although hospital admissions for ICH have increased by 18% in the past decade, mortality has not fallen (Qureshi et al, 2009). To date, no acute intervention beyond supportive medical care has been found to improve outcome in patients with ICH (Aronowski and Hall, 2005). Deterioration after ICH is caused not only by the space-occupying mass effects of the hematoma, but also by toxicity from blood components and subsequent perihematomal brain edema (Xi et al, 2006). Strategies aimed at limiting blood–brain barrier (BBB) dysfunction could be therapeutic targets for secondary injury.
Accumulating evidence suggests that blood-induced reactive oxygen species (ROS) have an important function in oxidative brain damage after ICH. In experimental ICH models, it seems that edema formation is related to BBB disruption from the toxic effects of red blood cell lysis, especially that of hemoglobin (Hb) (Xi et al, 2001; Huang et al, 2002). Hemoglobin is a major component of blood and a potent mediator of oxidative stress after ICH. Oxyhemoglobin in its conversion to methemoglobin releases superoxide (Misra and Fridovich, 1972; Winterbourn et al, 1976), and iron released from Hb causes lipid peroxidation and protein oxidation in vivo as well as in vitro (Regan and Panter, 1993; Qu et al, 2005).
Matrix metalloproteinases (MMPs) comprise a large family of zinc-endopeptidase that increases BBB permeability by degrading the extracellular matrix and tight junction proteins in endothelial cells (Rosenberg, 2009). MMPs, in particular MMP-9, have a pivotal function in brain edema after ICH. Knockout mice that lack the MMP-9 gene showed less edema than their wild-type (Wt) littermates in a model of ICH (Xue et al, 2006; Tejima et al, 2007). Oxidative stress is involved in MMP activation. In vitro, MMP-2 and MMP-9 can be activated by ROS, which oxidize a thiol bond responsible for their activation (Rajagopalan et al, 1996). Hemoglobin potently upregulated MMP-9 levels in an astrocyte culture, and a free radical scavenger reduced Hb-induced MMP-9 levels (Tejima et al, 2007). Moreover, oxidative stress accompanied by cerebral ischemia also facilitated an increase in MMP-9 activity (Gasche et al, 2001). Antioxidant enzymes have a critical function in the mechanism by which cells counteract the detrimental effects of ROS. Superoxide dismutase is one of these antioxidant enzymes. Copper/zinc-superoxide dismutase (SOD1)-deficient mice showed drastic MMP-9 activation after transient focal cerebral ischemia (tFCI) (Gasche et al, 2001). In this study, we examined the hypothesis that Hb-induced oxidative stress may contribute to MMP-9 activation and BBB dysfunction and lead to apoptosis in vivo. Furthermore, we also investigated the effect of SOD1 overexpression on Hb-induced oxidative damage, BBB dysfunction, and apoptosis.
Materials and methods
Animal Model
All animals were treated in accordance with Stanford University guidelines, and the animal protocols were approved by Stanford University's Administrative Panel on Laboratory Animal Care. Rat Hb (Sigma-Aldrich, St Louis, MO, USA) was reduced to oxyhemoglobin with a 10-fold molar excess of sodium dithionite as described earlier (Meguro et al, 2001). The concentration of the final product was determined using Drabkin's reagent. Hemoglobin solution was adjusted to 2 mmol/L (expressed as the concentration of the Hb tetramer), which is nearly the same concentration as that of whole blood. Male Sprague–Dawley rats (270 to 330 g; Charles River Laboratories, Wilmington, MA, USA) were anesthetized with 2.0% isoflurane in 68% nitrous oxide and 30% oxygen, and placed in a stereotactic frame. A 30-gauge needle attached to a 10-μL Hamilton microsyringe was inserted into the striatum via a polyethylene tube (3.5 mm lateral to the midline, 0.2 mm anterior to the coronal suture of the bregma, 5.5 mm below the surface of the skull, and then withdrawn 0.5 mm), and 2.5 μL of Hb was injected over 10 minutes using a microsyringe pump. As a control experiment, 2.5 μL of saline was infused in the same way. After infusion, the needle was left in place for 10 minutes and then slowly removed. The burr hole was sealed with bone wax and the incision was closed. Rectal temperature was controlled at 37 ± 5°C during surgery with a homeothermic blanket. Mean arterial blood pressure, pH, arterial oxygen, carbon dioxide tensions (pO2 and pCO2), and hematocrit were controlled within the normal range (mean arterial blood pressure, 80 to 120 mm Hg; pO2, 80 to 120 mm Hg; pCO2, 35 to 45 mm Hg; hematocrit, 38% to 43%). The animals were maintained at 20°C with ad libitum access to food and water.
Drug Treatment
To evaluate the involvement of MMPs in BBB disruption, the rats were treated with a nonselective MMP inhibitor (MMP-1, -3, -8, and -9 blocker), FN-439 (50 μg/μL of 4-Abz-Gly-Pro-D-Leu-D-Ala-NHOH in saline; EMD Chemicals, Gibbstown, NJ, USA), 30 minutes before the Hb injection, which was administered intraventricularly (10 μL, 1.5 mm lateral, 0.8 mm posterior, 3.6 mm deep to the bregma). The dose of FN-439 was determined according to an earlier study (Reeves et al, 2003), which showed that about 1 μmol of FN-439 injected intracerebroventricularly inhibited over 80% of MMP-9 activity in the brain.
Copper/Zinc-Superoxide Dismutase Transgenic Rats
Heterozygous SOD1 transgenic (Tg) rats with a Sprague–Dawley background carrying human SOD1 genes were derived from the founder stock and were further bred with Wt Sprague–Dawley rats to generate heterozygous rats as described earlier (Chan et al, 1998). There were no observable phenotypic differences in the brain vasculature between the SOD1 Tg rats and their Wt littermates (Chan et al, 1998).
Gelatin Zymography
To analyze gelatinase activity in the injured striata, gelatinases were first extracted from the brain tissue of the rats as described earlier (Kim et al, 2003) with some modification. The rats were perfused with phosphate-buffered saline (PBS) containing heparin, and then the injured striata were removed and frozen in liquid nitrogen 4, 8, 24, and 72 hours after the Hb or saline injection. The brain tissue from the striata was homogenized in lysis buffer (50 mmol/L Tris-HCl, pH 7.6, 150 mmol/L NaCl, 5 mmol/L CaCl2, 0.05% BRIJ-35, 0.02% NaN3, 1% Triton X-100 plus 1% protease inhibitor cocktail [Sigma-Aldrich]). The homogenates were centrifuged at 14000 g for 20 minutes. Protein concentrations were determined by the BCA protein assay method (Thermo Fisher Scientific, Waltham, MA, USA) using bovine serum albumin as a standard. The same amount of protein from each sample was incubated for 60 minutes with gelatin-Sepharose 4B (GE Healthcare, Piscataway, NJ, USA) with constant rotation. After incubation and centrifugation, the pellets were washed three times with equilibrium buffer (50 mmol/L Tris-HCl, pH 7.6, 150 mmol/L NaCl, 5 mmol/L CaCl2, 0.05% BRIJ-35, 0.02% NaN3) and were resuspended in 80 μL of elution buffer (equilibrium buffer with 10% dimethylsulfoxide) for 30 minutes. The supernatants were collected and then each sample was mixed with equal amounts of sodium dodecyl sulfate sample buffer (Invitrogen, Carlsbad, CA, USA). The samples were loaded on 10% sodium dodecyl sulfate-polyacrylamide electrophoresis gels with 0.1% porcine skin gelatin as a substrate (Invitrogen). After electrophoresis, the gels were incubated with renaturing buffer (Invitrogen) for 1 hour to remove the sodium dodecyl sulfate, and then incubated for 24 hours at 371C with developing buffer (Invitrogen). After incubation, the gels were stained for 60 minutes in Coomassie blue R-250 (0.25% Coomassie brilliant blue R-250, 50% methanol, 10% acetic acid) and destained in 30% methanol and 10% acetic acid until clear bands of gelatinolysis appeared on a blue background. A mixture of human MMP-2 and MMP-9 (CC073; Millipore, Billerica, MA, USA) was used as a gelatin standard. The images were scanned and quantification was performed using Multi-Analyst software (Bio-Rad Laboratories, Hercules, CA, USA).
In Situ Zymography and Immunofluorescence Staining
To examine in situ gelatinolytic activity, an in situ zymography was performed as described (Gasche et al, 2001) with some modification. The brains were removed without fixation, rapidly frozen in 2-methylbutane with dry ice. They were sectioned with a cryostat to a thickness of 10 μm. The sections were incubated with 40 μg/mL DQ gelatin conjugate (Invitrogen), a fluorogenic substrate, at 37°C for 2 hours and then, after washing, were fixed in 4% paraformaldehyde in PBS. DQ gelatin cleaved by MMPs resulted in a green fluorescent product (excitation, 495 nm; emission, 515 nm). To examine the cellular localization of the gelatinolytic activity, some fixed sections were incubated with a few primary antibodies specific for neurons (neuron-specific nuclear protein (NeuN), 1:100 dilution; Millipore) and astrocytes (glial fibrillary acidic protein, 1:400; Millipore). The sections were incubated overnight at 4°C, rinsed with PBS, and incubated for 2 hours at room temperature with a secondary antibody conjugated with Alexa 594 dye. Endothelial cells were detected by injecting Texas Red-conjugated Lycopersicon esculentum (tomato) lectin (0.5 mg; Vector Laboratories, Burlingame, CA, USA) via the jugular vein 30 minutes before the rats were killed. The sections were covered with VECTASHIELD mounting medium with 4′,6-diamidino-2-phenylindole (Vector Laboratories) and examined under an Axioplan 2 microscope (Carl Zeiss, Thornwood, NY, USA).
In Situ Detection of Reactive Oxygen Species Production
We investigated the production of superoxide by in situ detection of oxidized hydroethidine (HEt) as described earlier (Murakami et al, 1998). Hydroethidine (Invitrogen) is diffusible into the central nervous system parenchyma after an intravenous injection and is selectively oxidized to ethidium by ROS, especially superoxide. Hydroethidine solution (1 mL of 1 mg/mL in 1% dimethyl sulfoxide with saline) was administered intravenously 1 hour before the animals were killed. Oxidized HEt fluorescence was observed at an excitation of 510 nm and emission of >580 nm and quantified with ImageJ software (version 1.42q; NIH, Bethesda, MD, USA). For fluorescent double staining of the ethidium signal and the endothelial barrier antigen (EBA), anesthetized animals were perfused with 2 U/mL heparin PBS solution and subsequently with 4% formaldehyde in PBS solution. The brains were removed, postfixed for 24 hours, and sectioned at 50 mm with a vibratome. Sections were incubated with anti-EBA (1:400 SMI Monoclonal Antibodies; Covance, Princeton, NJ, USA) at 4°C overnight, followed by Alexa 594-conjugated anti-mouse immunoglobulin G (IgG) (Invitrogen) at room temperature for 2 hours. For fluorescent double staining of the ethidium signal and IgG, a sample was prepared as described in the in situ zymography method. Sections were incubated with Alexa 488-conjugated anti-rat IgG (1:500; Invitrogen) at room temperature for 2 hours. Slides were covered with 4′,6-diamidino-2-phenylindole and observed with a fluorescence microscope.
Measurement of Blood–Brain Barrier Disruption
Blood–brain barrier disruption was evaluated in the MMP inhibitor-treated rats (n = 5) and the SOD1 Tg rats. A sample was prepared as described in the gelatin zymography method. To measure serum protein (albumin) leakage, Western blot analysis was performed. After adding one-third volume of lithium dodecyl sulfate sample buffer (Invitrogen), we loaded 5 μg of protein per lane. Proteins were separated with the NuPAGE gel system (Invitrogen) and transferred to a polyvinylidene difluoride membrane (Invitrogen). The primary antibodies were a 1:20,000 dilution of anti-albumin (Bethyl Laboratories, Montgomery, TX, USA) and a 1:100,000 dilution of anti-β-actin (Sigma-Aldrich). Western blot analysis was performed with horseradish peroxidase-conjugated anti-sheep (Millipore) or anti-mouse secondary antibodies (Cell Signaling Technology, Beverly, MA, USA) using Supersignal West Pico and/or Femto substrate (Thermo Fisher Scientific). Images were scanned and the results were quantified using MultiAnalyst software (Bio-Rad).
Detection of Protein Oxidation
Samples were prepared as described using the gelatin zymography method. With the use of a commercial kit (Millipore), we observed the carbonyl groups as indicators of oxidative protein damage. The samples were incubated with 2,4-dinitrophenylhydrazone, and 2,4-dinitrophenylhydrazone-derivatized carbonyl groups were detected by Western blot analysis with an anti-2,4-dinitrophenylhydrazone antibody. The images were scanned and quantification was performed using Multi-Analyst software.
Cell Death Assay and In Situ Labeling of DNA Fragmentation
For quantification of apoptosis-related DNA fragmentation, we used a commercial enzyme immunoassay to determine cytoplasmic histone-associated DNA fragments (Roche Applied Science, Indianapolis, IN, USA) and to detect apoptotic but not necrotic cell death. The brain tissue from the striatum was homogenized in 20 mmol/L HEPES-KOH, pH 7.5, 10 mmol/L KCl, 1.5 mmol/L MgCl2, 1 mmol/L EDTA, and 1 mmol/L EGTA plus 1% protease inhibitor cocktails (Sigma-Aldrich). The homogenate was centrifuged at 10,000g for 10 minutes at 4°C. The supernatant was collected and 20 μg of protein was used for the enzyme-linked immunosorbent assay, according to the manufacturer's protocol. For in situ labeling of DNA fragmentation, the rats were deeply anesthetized and perfused with ice-cold heparinized saline followed by 4% paraformaldehyde in PBS. Their brains were removed and postfixed by immersion in the same fixative at 4°C overnight and then cryoprotected in 20% sucrose in PBS at 4°C. Frozen coronal sections (10 μm) at the level of the striatum were placed on slides and stained using terminal deoxynucleotidyl transferase-mediated uridine 5′-triphosphate-biotin nick end labeling (TUNEL) reaction to detect the DNA-free 3-OH ends as described earlier (Niizuma et al, 2008).
Quantification and Statistical Analysis
All data are presented as mean ± s.e.m. Statistical analyses were determined by Student's t test or by a standard one-way analysis of variance with Dunnett's posttest (InStat Software; GraphPad Software, San Diego, CA, USA). P values <0.05 were considered statistically significant.
Results
Reactive Oxygen Species Production Increased After Hemoglobin Injection
To confirm whether ROS occur in the striatum after Hb injection, the temporal and spatial production of ROS was investigated using a method of in situ detection of oxidized HEt. Oxidized HEt signals were increased in the cytosol of the striatum 1 hour after the Hb injection compared with saline-injected controls (Figures 1A and 1B). In the injured striata 4 and 8 hours after the Hb injection, oxidized HEt-positive cells were distributed more widely compared with the 1-hour sample (Figures 1C and 1D) and oxidized HEt signals were significantly increased compared with saline-injected controls (Figure 1E). These results indicate that superoxide is produced in the striatum after Hb injection from an early time point.
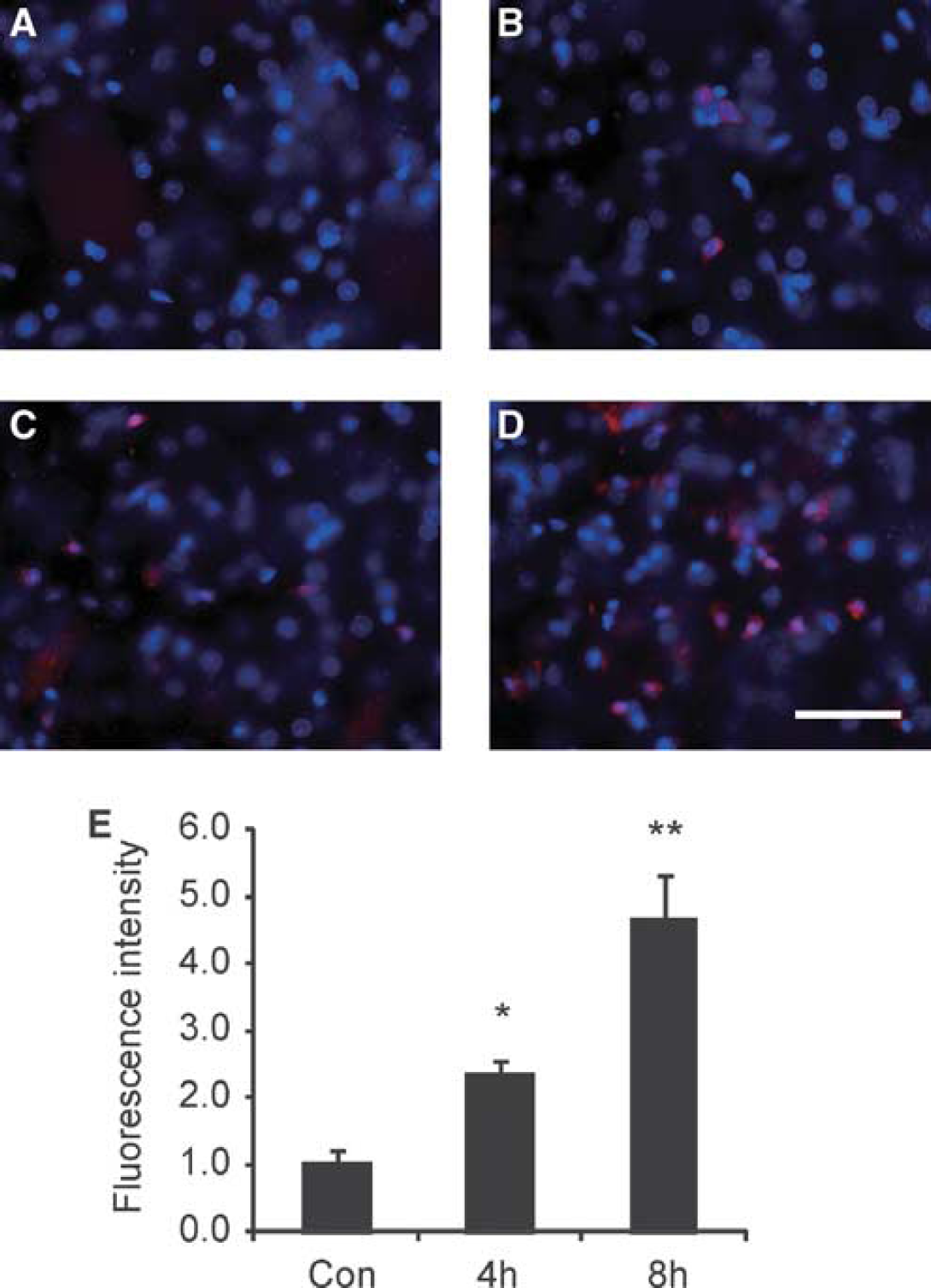
Changes in oxidized HEt signals (red) after the Hb injection. (
Matrix Metalloproteinase-9 Activity was Highly Upregulated After Hemoglobin Injection
To investigate MMP-9 levels after Hb injection, we performed gel zymography, which showed that MMP-9 was evident as double bands of 92 and 88 kDa molecular weight, which is consistent with earlier reports (Gursoy-Ozdemir et al, 2004; Kamada et al, 2007). Total MMP-9 levels (both bands together) were quantified. MMP-9 activity was slightly detectable in the sham-control brains, but began to increase by 4 hours after the Hb injection, peaked at 8 hours, then decreased by 24 hours, and declined to basal levels by 72 hours (Figures 2A and 2B). In contrast, MMP-2 was apparent as a single band of 72 kDa molecular weight in the sham-control brains and showed no significant change until 24 hours after the Hb injection. A significant but modest increase in MMP-2 levels was observed 72 hours after the Hb injection (Figures 2A and 2B). These results suggest that MMPs are upregulated in the striatum after Hb injection.
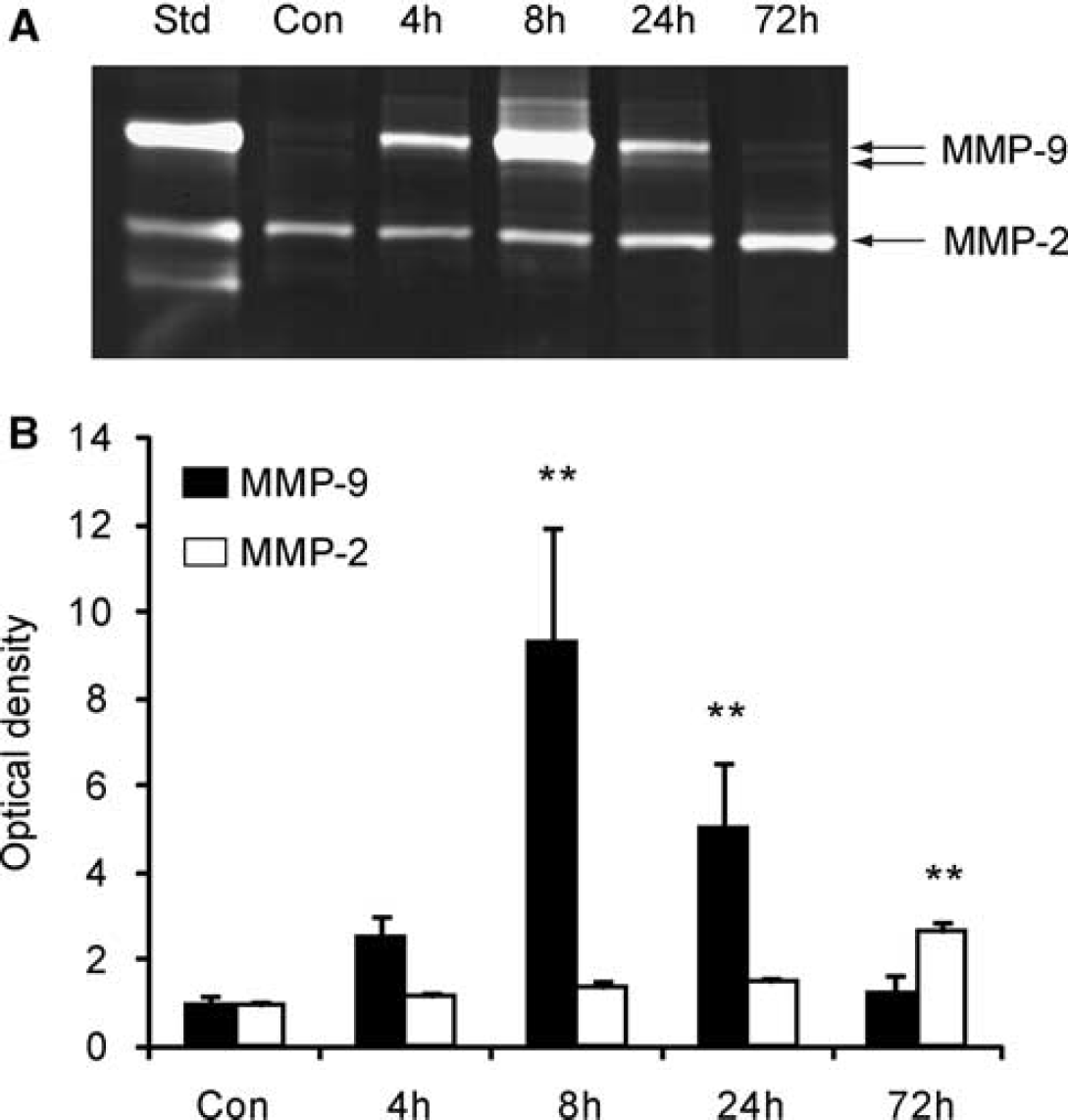
Changes in MMP levels after the Hb injection. (
In Situ Gelatinolytic Activity Mainly Localized in Neurons and Vessel Walls
To confirm the cellular localization of gelatinolytic activity after Hb injection, we performed double immunofluorescence for in situ zymography and cellular markers. Most of the gelatinolytic activity was detectable in neurons and vessel walls in the striatum after the Hb injection, but was very slight in the saline-injected controls (data not shown). In some cells, gelatinolytic activity was superimposed on the nuclei of NeuN-positive neurons (Figure 3A), which is also confirmed in studies of FCI (Gasche et al, 2001) and spontaneous ICH (Wakisaka et al, 2010). Colocalization between in situ gelatinolysis and astrocytic end-feet at the vessel wall was observed, whereas no gelatinolysis was detectable in the cellular body of the astrocytes (Figure 3B). Colocalization of in situ gelatinolytic activity and lectin, a marker of endothelial cells, was observed on the vessel walls (Figure 3C). These results indicate that gelatinolytic activity is triggered in vessel walls and brain parenchymal cells after Hb injection.
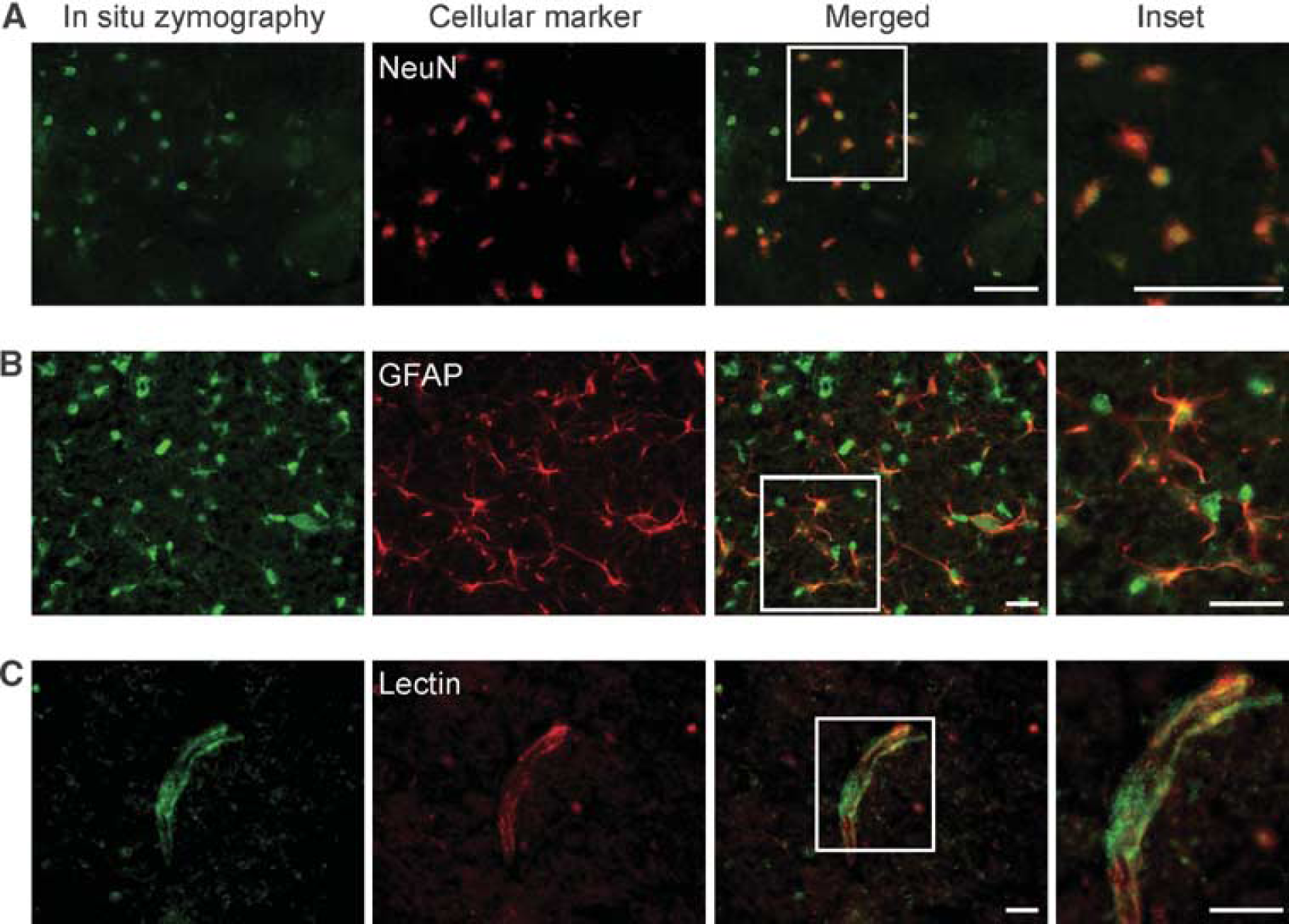
Representative photomicrographs of double staining for in situ zymography and different cell markers (
Increased Hydroethidine Signals were Accompanied by In Situ Gelatinolysis and Immunoglobulin G Leakage
As we have previously shown that in situ gelatinolytic activity also localizes where oxidized HEt signals increase after tFCI (Gasche et al, 2001) and spinal cord injury (Yu et al, 2008), we performed in situ zymography on cryosections obtained from brain tissue injected with HEt. Double immunofluorescence for in situ zymography and HEt showed that gelatinolytic activity colocalized with oxidized HEt signals on the morphologically identified vessel walls of the striata 8 hours after the Hb injection (Figures 4B and 4C). To confirm whether the BBB was compromised after Hb injection, we performed immunofluorescence for IgG, the leakage of which was detected in the injured striata 8 hours after the Hb injection (Figures 4G and 4H). In contrast, IgG leakage was detected neither in the corresponding contralateral region (Figures 4E and 4F) nor in the saline-injected control (data not shown). To investigate the relationship between IgG extravasation and ROS production, we also performed double immunofluorescence for IgG and HEt, which showed that IgG leakage occurred around vessels in which oxidized HEt signals were increased (Figures 4J and 4K). These results imply that Hb-induced oxidative stress is implicated in increased gelatinolytic activity and IgG leakage on vessel walls.
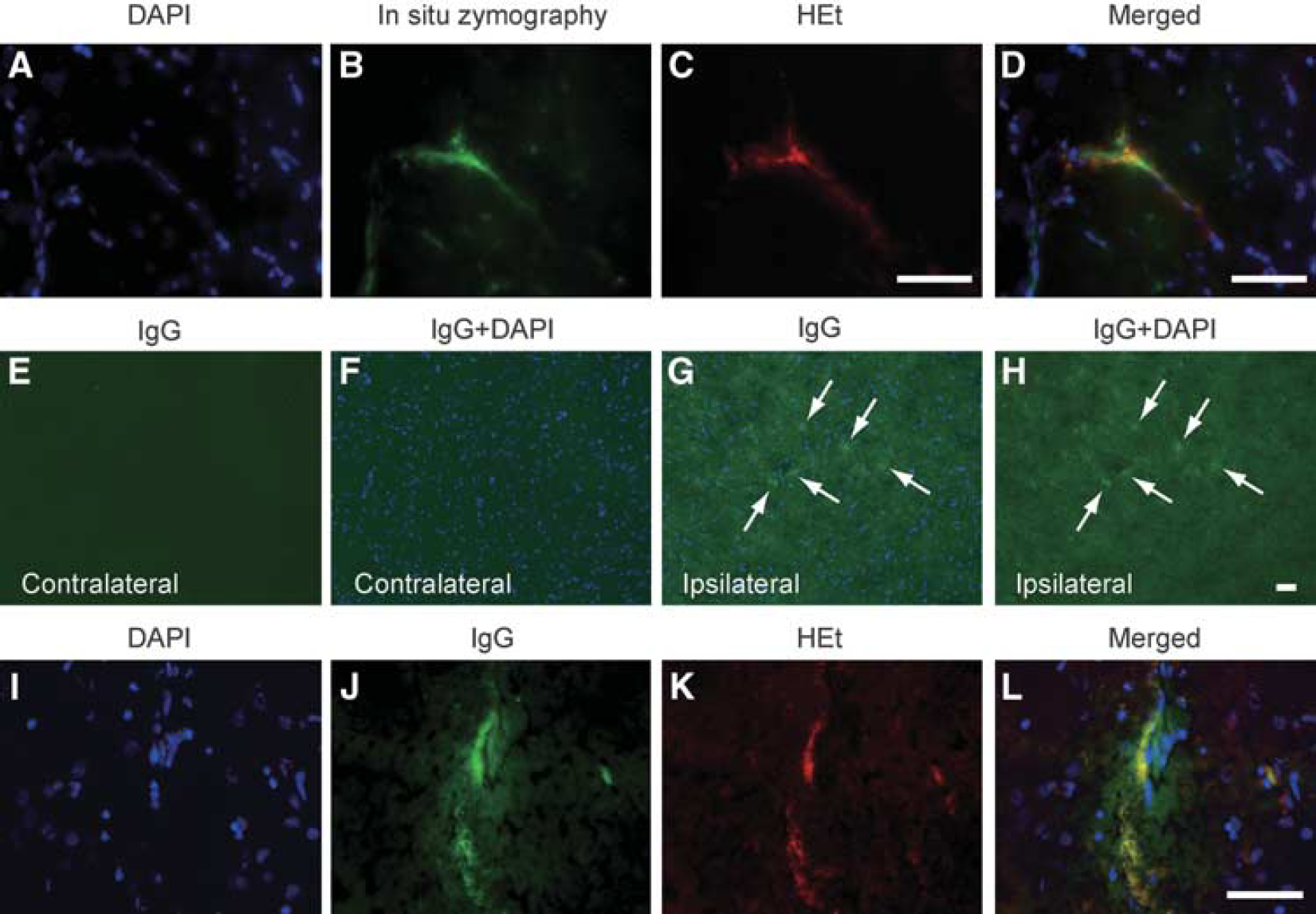
Increased HEt signals accompanied by in situ gelatinolysis or IgG leakage. (
Endothelial Barrier Antigen Immunofluorescence was Decreased After Hemoglobin Injection
Endothelial barrier antigen is an endothelial protein found in areas with blood–brain or blood–nerve barriers. Pathological conditions that include BBB disruption and leakage of plasma proteins are associated with reduced EBA expression in brain endothelial cells (Gursoy-Ozdemir et al, 2004; Lu et al, 2008). To examine EBA immunoactivity in brain microvessels in the striata after Hb injection, we immunostained EBA protein on cryosections obtained from brain tissue injected with HEt. Double immunofluorescence showed that EBA was decreased and EBA-positive microvessels were degraded to short segments in the injured striata, in which oxidized HEt signals were increased (Figures 5E to 5H), compared with the corresponding contralateral region (Figures 5A to 5D) or saline-injected control (data not shown). This result also indicates that the BBB might be altered in the striatum after Hb injection.
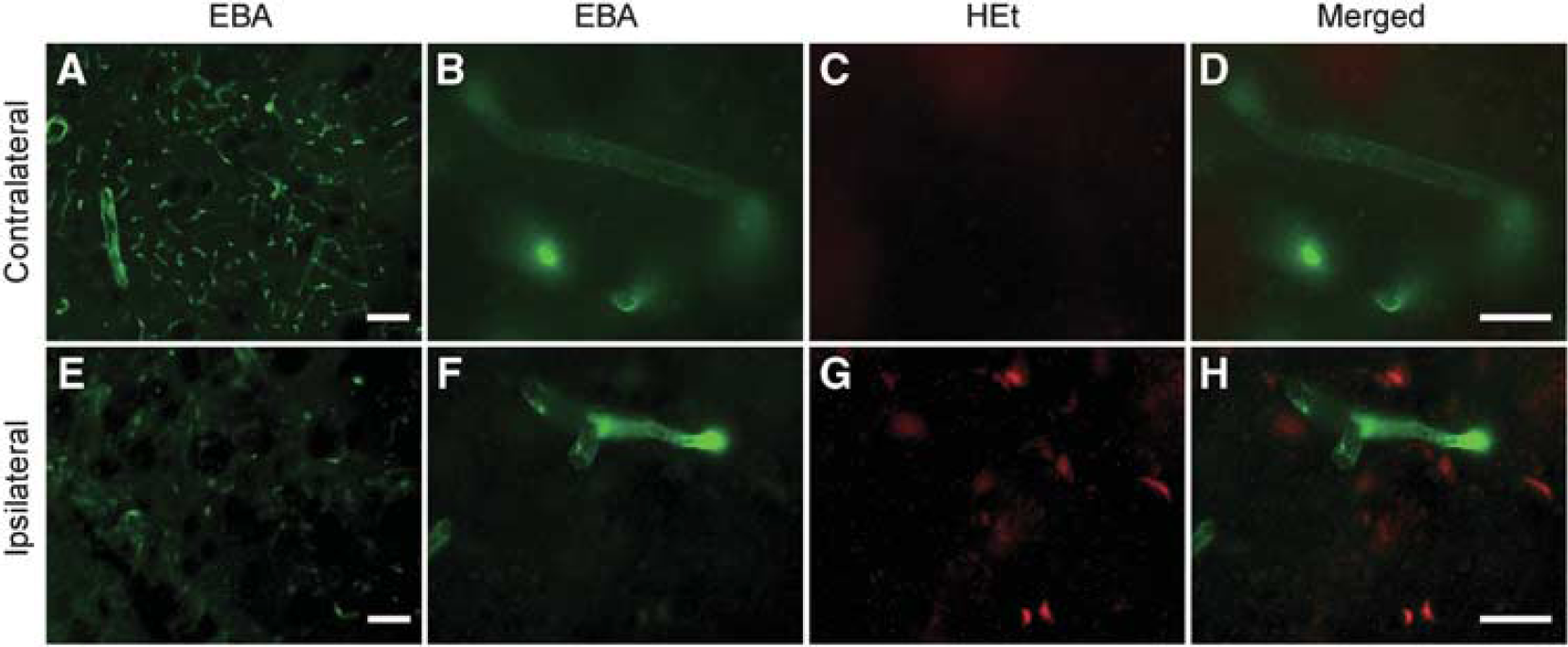
Representative photomicrographs of double staining for EBA and oxidized HEt. EBA-positive microvessels were degraded to short segments in injured striata (
Matrix Metalloproteinase-9 is Associated with Albumin Leakage After Hemoglobin Injection
Our results show that increased HEt signals colocalized with increased gelatinolytic activity and IgG extravasation, and decreased EBA immunoactivity. These results suggest a strong relationship between increased gelatinolytic activity and BBB dysfunction. To address the role of increased gelatinolytic activity on Hb-induced albumin leakage, we compared albumin leakage between rats treated with the MMP inhibitor, FN-439, or the vehicle 8 hours after the Hb injection. First, we tested whether administration of FN-439 reduced Hb-induced gelatinase levels. MMP-9 levels were significantly inhibited in FN-439-treated animals compared with vehicle-treated animals, as assessed by gel zymography (Figures 6A and 6B). There was no difference in MMP-2 levels between these two groups of animals (Figure 6A). We then evaluated albumin leakage by Western blot analysis, which showed that FN-439 significantly inhibited albumin leakage 8 hours after the Hb injection (Figures 6C and 6D). In the samples of the vehicle-treated or naive animals, no albumin leakage was observed (Figure 6C). These results suggest that an increased MMP-9 level is associated with BBB dysfunction.
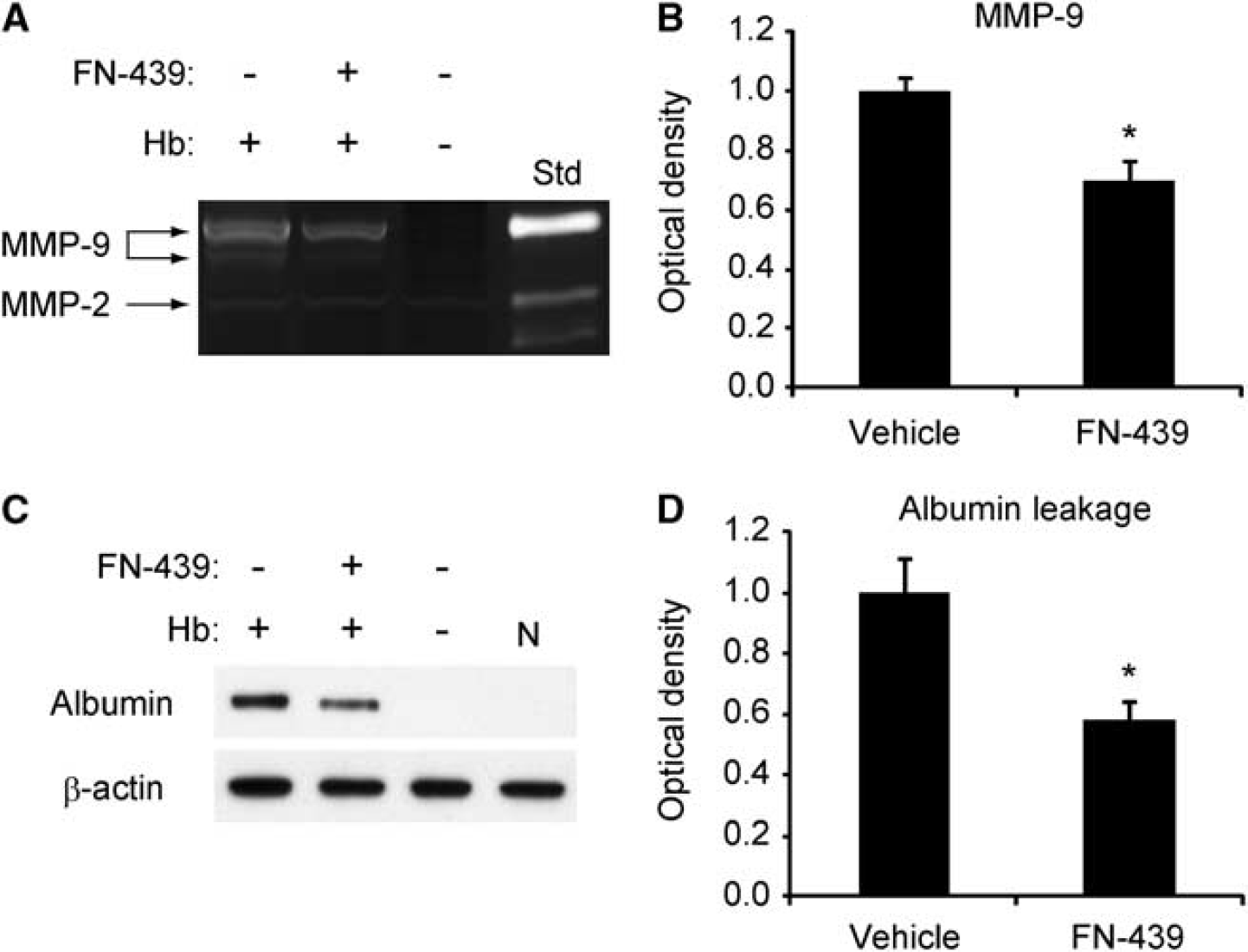
Effect of the nonselective MMP inhibitor, FN-439, on gel zymography and BBB permeability 8 hours after the Hb injection. (
Copper/Zinc-Superoxide Dismutase Overexpression Protects Against Albumin Leakage and Apoptotic Cell Death After Hemoglobin Injection
To confirm that ROS production is associated with increased MMP-9 levels and BBB dysfunction, we investigated albumin leakage after Hb injection using SOD1 Tg rats and their Wt littermates. First, we investigated whether SOD1 overexpression reduced ROS production 8 hours after Hb injection. Enhanced oxidized HEt signals were observed in the striata of the Wt rats 8 hours after the Hb injection, but were significantly decreased in the SOD1 Tg rats (Figure 7A). To further examine whether Hb-induced oxidative stress was decreased in the SOD1 Tg rats after Hb injection, the level of the carbonyl groups was determined by Western blot analysis. An increase in the level of the carbonyl groups was observed in a time-dependent manner after the Hb injection in both Wt and SOD1 Tg rats. However, their level in the SOD1 Tg rats was significantly decreased compared with the Wt rats both 8 and 72 hours after the Hb injection (Figures 7B and 7C). We then examined by gel zymography whether MMP-9 levels were reduced in the SOD1 Tg rats. It showed that MMP-9 levels were decreased compared with the Wt rats 8 hours after the Hb injection (Figure 7D). There was no difference in MMP-2 levels between the SOD1 Tg and Wt rats (Figure 7D). Finally, we investigated whether albumin leakage was reduced in the SOD1 Tg rats after Hb injection. Western blot analysis showed that it was significantly attenuated compared with the Wt rats 8 hours after the Hb injection (Figure 7E).
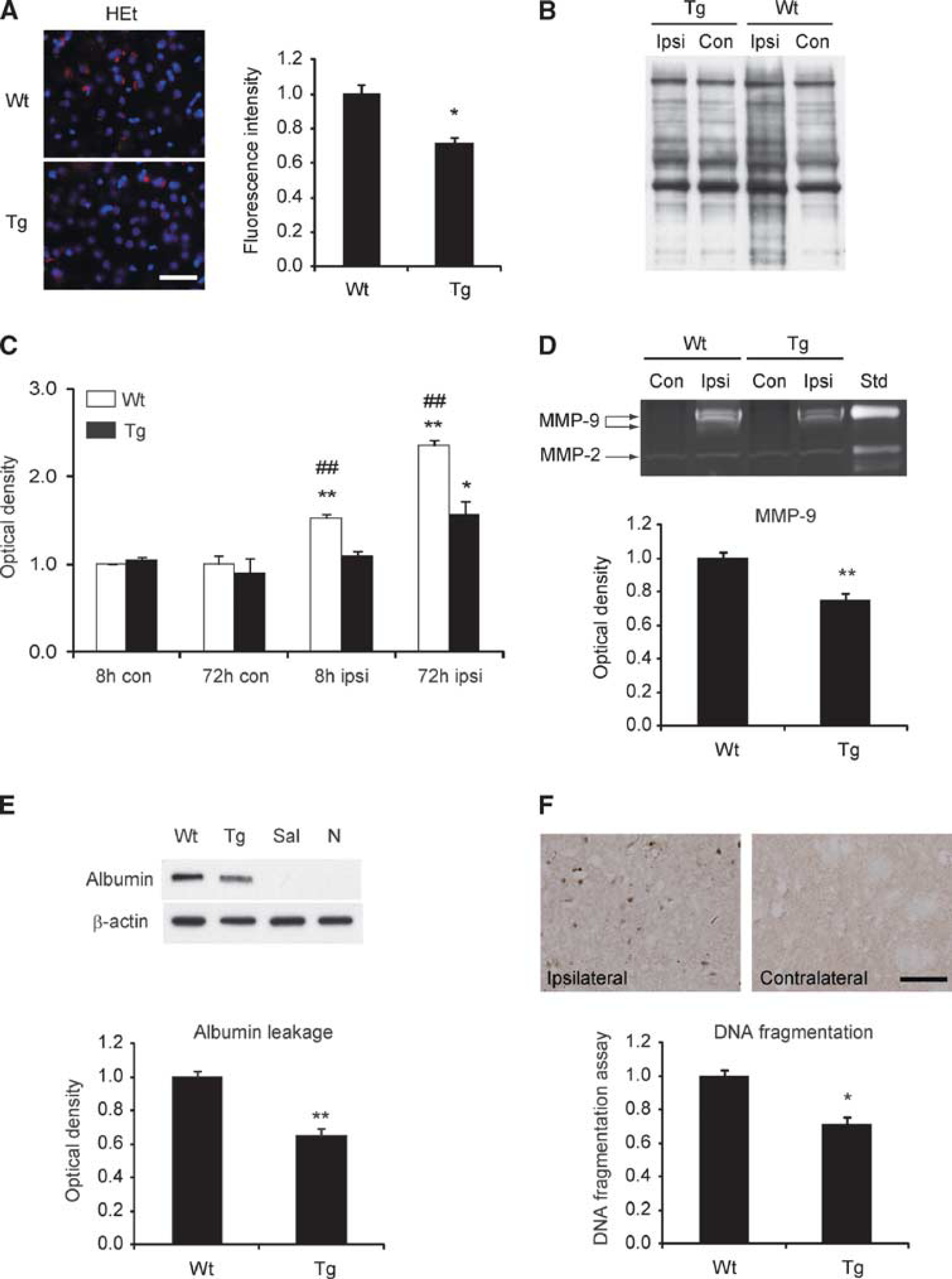
Oxidized HEt, MMP-9 levels, BBB disruption, and DNA fragmentation assay in SOD1 Tg rats 8 hours after the Hb injection. (
Oxidative stress is known to have extensive interaction with apoptosis. To confirm whether apoptosis occurs after Hb injection, DNA fragmentation was analyzed with in situ TUNEL and a commercial enzyme immunoassay. The TUNEL-positive cells were detected in the striata 24 hours after the Hb injection, whereas no TUNEL-positive cells were observed in the contralateral region (Figure 7F). We then examined apoptosis-related DNA fragmentation using the commercial enzyme immunoassay. DNA fragmentation in the striata 72 hours after the Hb injection was significantly decreased in the SOD1 Tg rats compared with the Wt rats. Taken together, these results indicate that SOD1 overexpression attenuates ROS production, MMP-9 levels, BBB dysfunction, and subsequent apoptotic cell death.
Discussion
Accumulating evidence suggests that ROS have a critical function in oxidative brain damage after ICH. Our current data show that oxidative stress has an important function in Hb-induced brain injury in vivo. In addition, our data suggest that MMPs contribute to BBB dysfunction in this pathophysiological process. We base these conclusions on the following findings. First, early MMP-9 levels after the Hb injection were parallel to increased HEt signals. In situ gelatinolytic activity colocalized with increased HEt signals in the vessel walls. Second, increased HEt signals were accompanied by IgG leakage and a decrease in immunoactivity of EBA in the vessel walls. Third, administration of the MMP inhibitor prevented increased MMP-9 levels and albumin leakage. Finally, reduction in oxidative stress by SOD1 overexpression inhibited albumin leakage along with increased MMP-9 levels, and subsequent apoptotic cell death.
The role of ROS as a mediator of oxidative brain damage after ICH is well known. Cell culture studies have shown that Hb-induced neurotoxicity is mediated by iron released from Hb, as free radical scavengers and an iron chelator completely blocked Hb-induced neurotoxicity (Regan and Panter, 1993; Wang et al, 2002). Indeed, ROS production was detected after exposure to Hb in vitro (Regan and Rogers, 2003). Moreover, the level of the carbonyl groups as an index of tissue protein oxidation was increased after the Hb injection in vivo (Qu et al, 2005). In this study, ROS production was markedly increased in the injured striatum as early as 1 hour after the Hb injection and the increased level of the carbonyl groups was also observed 24 hours after the Hb injection. Furthermore, increased ROS production and the carbonyl groups generated by Hb were attenuated by SOD1 overexpression. This attenuation is supported by reports showing that oxyhemoglobin in its conversion to methemoglobin releases superoxide (Misra and Fridovich, 1972; Winterbourn et al, 1976). Taken together, these results suggest that Hb can be a potent pro-oxidant and a source of ROS in vivo.
MMPs comprise a large family of zinc-endopeptidases that increases the permeability of the BBB by degrading the extracellular matrix and tight junction proteins in endothelial cells (Rosenberg, 2009). MMPs, especially MMP-9, have a critical function in proteolytic degradation of the BBB in the pathological process. Knockout mice that lack the MMP-9 gene showed less edema than their Wt littermates in a model of ICH (Xue et al, 2006; Tejima et al, 2007), as well as in cerebral ischemia (Asahi et al, 2001). In this study, we showed that increased gelatinolytic activity colocalized with the endothelial marker at the vessel walls, and that IgG leakage was observed around the vessels in the injured striata. Furthermore, treatment with the nonspecific MMP inhibitor, FN-439, significantly decreased Hb-induced albumin leakage along with MMP-9 levels. Although loss of membrane integrity is probably caused directly by ROS-induced lipid peroxidation that can be catalyzed by iron released from Hb (Regan and Panter, 1993; Qu et al, 2005), these results suggest that MMPs are also implicated in Hb-induced BBB dysfunction, at least in part, through MMP-9 activation. After the Hb injection, we also observed a decrease in immunoactivity of EBA, which is a marker of BBB integrity. Targeting EBA by administration of the EBA antibody leads to opening of the BBB via paracellular and transcellular routes in vivo (Ghabriel et al, 2002). Our results showed that EBA-positive microvessels were degraded to short segments in the injured striata, in which oxidized HEt signals were increased, suggesting that the BBB might be altered in the injured striata after the Hb injection. This result is compatible with a study showing that EBA-immunoactive microvessels were decreased after focal ischemia/reperfusion through MMP activation, and that FN-439 inhibited the reperfusion-related decrease in EBA immunoactivity (Lu et al, 2008).
Reactive oxygen species, especially superoxide anions, have been implicated in activation and induction of MMP-9 in vivo as well as in vitro (Rajagopalan et al, 1996; Gu et al, 2002; Liu and Rosenberg, 2005). In this study, we showed that gelatinolytic activity colocalized with ROS production at the vessel walls. In addition, MMP-9 levels were paralleled by ROS production. These results indicate that Hb-induced ROS may also be involved in the induction and activation of MMPs. Reduction in oxidative stress in SOD1 Tg rats attenuated MMP activation after tFCI (Kamada et al, 2007), spinal cord injury (Yu et al, 2008), cold injury-induced brain trauma (Morita-Fujimura et al, 2000), and 3-nitropropionic acid injury (Kim et al, 2003). In contrast, SOD1-deficient mice or manganese-superoxide dismutase (SOD2) heterozygous mice with 50% SOD2 activity showed drastic MMP-9 activation after tFCI (Gasche et al, 2001; Maier et al, 2006). In this study, we showed that overexpression of SOD1 decreased oxidative stress and MMP-9 levels after the Hb injection, which might lead to decreased BBB dysfunction. These results are partly supported by a study showing that Hb potently upregulates MMP-9 levels in an astrocyte culture, and that a free radical scavenger reduces Hb-induced MMP-9 levels (Tejima et al, 2007). A few speculations can be provided for the attenuation of Hb-induced MMP-9 activation. SOD1 overexpression might attenuate MMP-9 activation by directly reducing oxidation of a thiol bond responsible for its activation (Rajagopalan et al, 1996). SOD1 overexpression might have an effect on redox-sensitive factors. MMP-9 synthesis is triggered by activation of redox-sensitive transcription factors such as nuclear factor-κB and activator protein-1 (Liu and Rosenberg, 2005). Indeed, nuclear factor-κB is rapidly activated in the perihematomal brain after ICH (Hickenbottom et al, 1999). We have reported that SOD1 overexpression attenuates acute activation of activator protein-1 after tFCI (Huang et al, 2001a) and downregulates nuclear factor-κB expression after tFCI (Huang et al, 2001b). Reduction in ROS by SOD1 overexpression might decrease with the induction of nuclear factor-κB and activator protein-1 and lead to reduced induction and activation of MMP-9.
In this study, a reduction in oxidative stress by SOD1 overexpression resulted in decreasing apoptotic cell death. Oxidative stress is known to have extensive interaction with apoptosis (Niizuma et al, 2009). It has been shown that Hb activates a caspase pathway and induces apoptosis in cultured cortical neurons (Wang et al, 2002) and cultured microvessel endothelial cells (Meguro et al, 2001). Meanwhile, extracellular activation of MMP-9 by nitric oxide can degrade the extracellular matrix laminin and contribute to apoptosis-like death (Gu et al, 2002, 2005). In addition, gelatinolytic activity in the nuclei of neurons promotes neuronal apoptosis after cerebral ischemia by degrading DNA repair enzymes, PARP-1 and XRCC-1 (Yang et al, 2010). Although we cannot determine whether caspase or MMP activation contributes to apoptotic cell death, based on existing data, it is clear that SOD1 overexpression is protective against apoptotic cell death after Hb injection. Cytochrome c release from the mitochondria after Hb injection is probably the underlying mechanism for apoptotic cell death (Matz et al, 2001). However, the cellular source (neurons, endothelia, astrocytes, microglia, etc.) was not determined in this study.
In conclusion, our study may suggest that oxidative stress has an important function in Hb-induced BBB dysfunction and subsequent cell death. More importantly, our results may indicate that SOD1 overexpression can attenuate the pathophysiological process through reduction of MMP activity. Reducing oxidative stress, thereby inhibiting MMP activation, could be a therapeutic target for secondary injury that accompanies brain hemorrhage.
Footnotes
Acknowledgements
The authors thank Liza Reola and Bernard Calagui for technical assistance and Cheryl Christensen for editorial assistance.
The authors declare no conflict of interest.