
Abstract
Osteopontin (OPN), a large secreted glycoprotein with an arginine, glycine, aspartate (RGD) motif, can bind and signal through cellular integrin receptors. We have shown previously that OPN enhances neuronal survival in the setting of ischemia. Here, we sought to increase the neuroprotective potency of OPN and improve the method of delivery with the goal of identifying a treatment for stroke in humans. We show that thrombin cleavage of OPN improves its ability to ligate integrin receptors and its neuroprotective capacity in models of ischemia. Thrombin-cleaved OPN is a twofold more effective neuroprotectant than the untreated molecule. We also tested whether OPN could be administered intranasally and found that it is efficiently targeted to the brain via intranasal delivery. Furthermore, intranasal administration of thrombin-treated OPN confers protection against ischemic brain injury. Osteopontin mimetics based on the peptide sequences located either N or C terminal to the thrombin cleavage site were generated and tested in models of ischemia. Treatment with successively shorter N-terminal peptides and a phosphorylated C-terminal peptide provided significant neuroprotection against ischemic injury. These findings show that OPN mimetics offer promise for development into new drugs for the treatment of stroke.
Introduction
Osteopontin (OPN) is a multifunctional acidic phosphoprotein found in all body fluids. It is expressed by macrophages and endothelial cells and found in the central nervous system (CNS). Although it is not a normal component of the extracellular matrix, OPN is a soluble factor that can mimic extracellular matrix signaling (Denhardt et al, 2001b). It contains an RGD (arginine, glycine, aspartate) motif, which binds an array of integrins that affect a wide range of cellular processes, such as adhesion, migration, differentiation, survival, and repair (Barry et al, 2000; Denhardt et al, 2001a, 2001b; Smith and Giachelli, 1998; Yokosaki et al, 1999). In the CNS, OPN is expressed at low basal levels in neurons (Shin et al, 1999) and microglia (Wang et al, 1998). During pathologic states, it is expressed by infiltrating macrophages (Wang et al, 1998). Most CNS cells express integrin receptors that permit interaction with OPN (Ellison et al, 1998; Gary and Mattson, 2001; Giancotti and Ruoslahti, 1999; Wang et al, 2001).
Activation of integrin receptors on neurons promotes cell survival and confers protection from ischemic injury (Gary et al, 2003). This has been shown using ligands for integrin receptors that are components of the extracellular matrix, such as laminin and fibronectin (Gary et al, 2003; Mosher, 2001). Recently, we showed that OPN is neuroprotective in models of ischemic stroke, and that protection is dependent on the activation of mitogen-activated protein kinase and phosphoinositide-3 kinase signaling pathways (Meller et al, 2005). The neuroprotective effect of OPN in vitro is lost in the presence of a short RGD-containing peptide, which acts as a competitive antagonist and blocks interactions with integrin receptors (Meller et al, 2005). This suggests that OPN also confers protection via integrin receptors similar to laminin and fibronectin. These three molecules share a classical RGD integrin-binding sequence. However, OPN contains a unique motif comprised of the sequence SLAYGLR (mouse) or SVVYGLR (human) that surrounds the RGD sequence (Green et al, 2001; Yamamoto et al, 2003). This motif increases the repertoire of integrins available for OPN to bind. Like laminin and fibronectin, OPN binds to the integrins αvβ3, αvβ1, αvβ5, α8β1, and α5β1 via the RGD sequence, but OPN can also bind integrins α4β1, α4β7, and α9β1 via the SLAYGLR motif. Importantly, the SLAYLGR motif is cryptic and unable to bind to integrins when in the native form of OPN. However, cleavage by thrombin at residue Arg153 (Arg168 in humans) exposes a C-terminal carboxylic acid required for binding to α4 and α9 integrins (Green et al, 2001). Thrombin cleavage of OPN may also improve the ability of integrin receptors to bind to the RGD sequence (Senger et al, 1994). The orientation of the RGD sequence, as determined by the surrounding residues, is a key element for favorable integrin binding and signaling (Ochsenhirt et al, 2006). Thrombin cleavage of OPN may provide an optimal orientation of the RGD sequence for integrin signaling. Thus, thrombin cleavage of OPN represents a potential means to improve the neuroprotective potency of OPN.
Intranasal administration is a noninvasive means of delivering therapeutics to the brain, bypassing the blood—brain barrier. Intranasal administration achieves efficient delivery of various proteins (e.g., insulin-like, fibroblast, and nerve growth factors) and other molecules (DNA plasmids and flavenoids) to the brain in animal models and humans (Born et al, 2002; Jin et al, 2002; Thorne and Frey, 2001). This approach has been used successfully with several proteins and peptides in the treatment of CNS diseases or injury (Born et al, 2002; Jin et al, 2002; Liu et al, 1992). Thus, intranasal administration offers a potential means of improved delivery of OPN to the brain.
There are limitations to the clinical application of proteins as drugs. These include rapid degradation of the protein by the action of specific and nonspecific proteases and poor absorption and transportation because of high molecular weight. Conformational flexibility can lead to undesirable side effects as multiple receptors can be activated. A means of circumventing these limitations is to explore structure—activity relationships with synthetic peptides, which can then be used for the rational design of peptidomimetics. We have tested synthetic peptides derived from the sequence of thrombin-treated OPN as an initial and requisite step to the rational design of OPN peptidomimetics.
Thus, we describe a means to enhance the neuroprotective potency of OPN (thrombin cleavage) and an improved method of targeting OPN to the brain via intranasal administration. Further, we have begun to explore the neuroprotective potential of peptidomimetic precursors, based on the sequence of thrombin-cleaved OPN.
Materials and methods
Peptides
Osteopontin peptides were custom-synthesized by Invitrogen (Carlsbad, CA, USA). Phosphorylated residues are indicated by: S(p) or T(p). A schematic diagram of OPN and the location of peptides NT (N-terminal) 109 to 153 and CT (C-terminal) 154 to 198 within the full-length sequence are shown in Figures 1A and 1B. Peptide sequences are as follows:
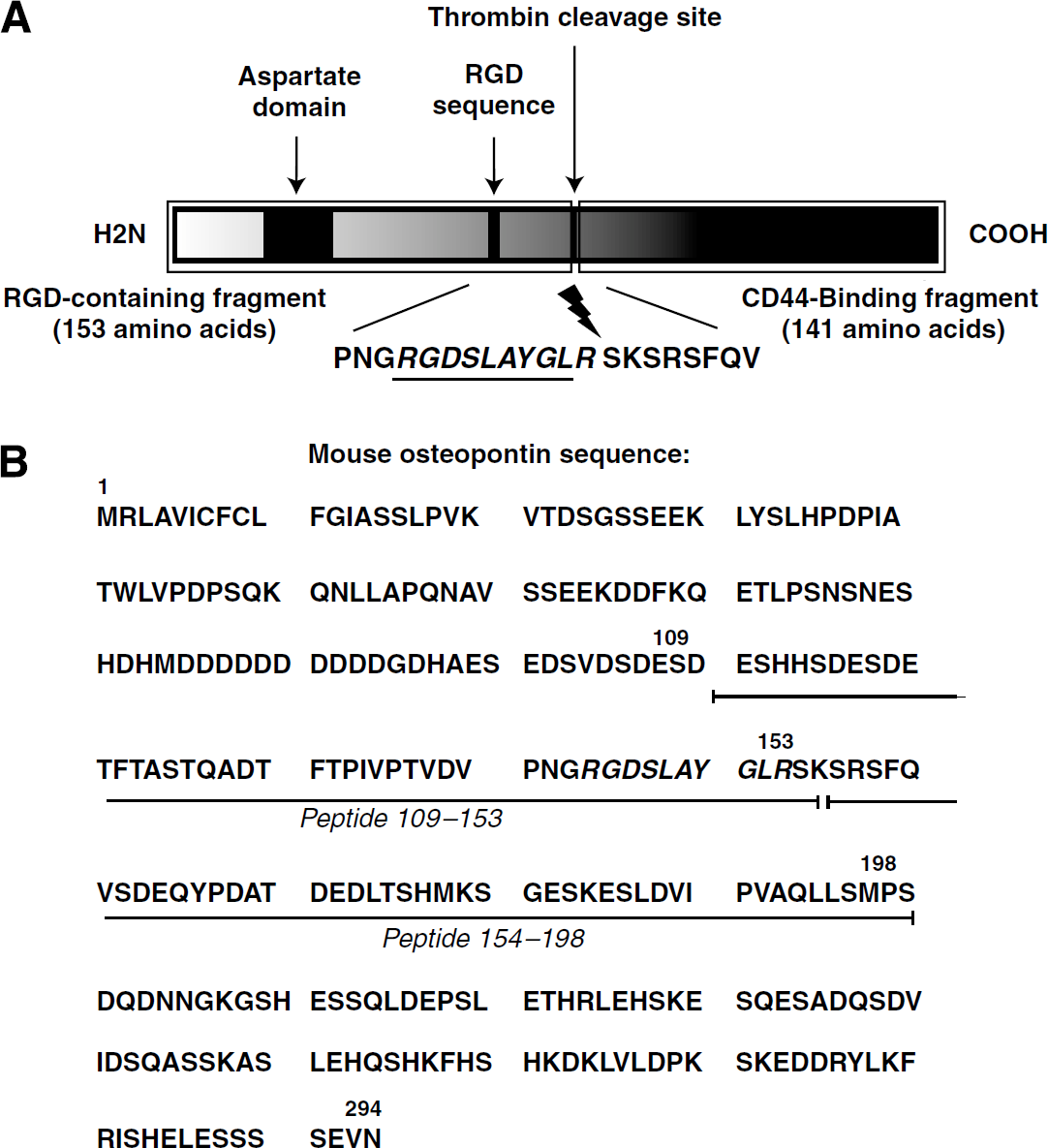
(
S(p)DES(p)HHS(p)DES(p)DETVTASTQADTFTPIVPTVDVPNGRGDSLAYGLR
SDESHHSDESDETVTASTQADTFTPIVPTVDVPNGRGDSLAYGLR
ASTQADTFTPIVPTVDVPNGRGDSLAYGLR
IVPTVDVPNGRGDSLAYGLR
RGDSLAYGLR
SKSRSFQVSDEQYPDAT(p)DEDLTS(p)HMKS(p)GESKESLDVIPVAQLLSM
SKSRSFQVSDEQYPDATDEDLTSHMKSGESKESLDVIPVAQLLSM
NVPGDVVITPDSEHSGLDSRAAQTSSEDHSGAYRLFPDTTTVDET
Thrombin Cleavage of Osteopontin
Recombinant mouse OPN (800 μL at 100 μg/mL; EMD Biosciences/Calbiochem, San Diego, CA, USA) was mixed with thrombin agarose beads (100 μL, Thrombin CleanCleave Kit; Sigma, Saint Louis, MO, USA) for 72 h at 37°C with shaking. Thrombin was removed by centrifugation (500g for 5 mins) and the supernatant was collected. The supernatant was further purified via passage through a 0.2 μm filter, which removed any remaining thrombin agarose beads (diameter of 45 μm) and allowed OPN and its cleaved fragments to pass through. Thrombin was covalently bound to the agarose beads with no detectable leakage of thrombin from the beads with a detection limit of <5 pmol/mL; thus, the cleaved fragments were not contaminated with residual thrombin.
Adhesion Assay with HEK 293 Cells
Ninety-six-well plates were coated with recombinant mouse OPN (thrombin-treated or untreated) at 2 μg/mL in phosphate-buffered saline (PBS) overnight at 4°C. Plates were washed three times with PBS and blocked with 200 μL/well of PBS-0.5% bovine serum albumin at room temperature for 1 h. HEK 293 cells were plated in adhesion buffer (PBS, 50 mmol/L HEPES, 0.1% bovine serum albumin, 0.1% glucose, and 0.05 mmol/L MnCl2) at a density of 1 × 105 cells/well. Cells were incubated at 37°C for 45 mins and then washed in PBS to remove nonadherent cells. Sigma FAST p-nitrophenylphosphate (50 μL) was added to each plate for 2 h at 37°C and plates were read at 405 nmol/L. The percentage of cells remaining in each well (adherent cells) was calculated using a standard curve generated from a control plate containing known quantities of cells in each well ranging from 0 to 250,000. The experiment was performed three times and the results averaged.
Neuronal Cell Culture
Cortical neuronal cultures were prepared from 1-day-old Sprague Dawley rat pups using the method of Goslin as described previously (Stenzel-Poore et al, 2003). Briefly, cortices were dissected from 10 to 12 rat pups, dissociated with papain (Worthington Biochemicals, Lakewood, NJ, USA), and grown in Neurobasal-A/B27/Glutamax media (Invitrogen) for 7 days. Before use, cells were plated on coverslips at a density of 1,000,000 cells per coverslip or in 96-well plates at a density of 100,000 cells/well.
Oxygen and Glucose Deprivation
Neuronal cells were washed with PBS (0.5 mmol/L CaCl2 and 1.0 mmol/L MgCl2; pH 7.4) and exposed to anaerobic conditions (85% N2, 5% H2, 10% CO2) for 120 mins at 37°C using an anaerobic chamber (Coy Laboratory Products Inc., Grass Lake, MI, USA). Oxygen—glucose deprivation (OGD) was terminated by replacement with growth media and return to a normoxic incubator. Cells were incubated with mouse recombinant OPN or peptide fragments thereof for 24 h after 120 mins exposure to OGD conditions.
Cell Viability Assays
For propidium iodide cell viability assays, coverslips containing cortical cells were incubated with propidium iodide (1.5 μg/mL) for 2 mins, washed with PBS, and fixed with 10% formalin. Cells were permeabilized with 0.1% Triton X-100 and then mounted onto glass slides using Vectashield mounting medium containing 4′,6′-diamidino-2-phenylindole (DAPI) (Vector Labs, Burlingame, CA, USA). Cells were treated with thrombin-treated (5 nmol/L) or untreated OPN (5 nmol/L) for 24 h after 120 mins OGD (Figure 3A) or with 5 nmol/L peptide CT 154 to 198 (p) and varying concentrations (0.1 to 10 μg/mL) of CD44-neutralizing antibody (BD Biosciences, San Jose, CA, USA) for 24 h after 120 mins OGD (Figure 5B). Cell viability was determined as the ratio of propidium iodide-stained cells to the total number of DAPI-stained cells, visualized using a fluorescence microscope (Leica GMBH, Bannockburn, IL, USA). Three fields of view were counted per slide and there were two slides per treatment per experiment (n2).
Thrombin-treated osteopontin (OPN) is more effective than untreated OPN at reducing neuronal death in response to ischemia. (
Peptides of osteopontin (OPN) are neuroprotective in vitro. (
For MTT cell viability assays, cells in 96-well plates were treated with 5 nmol/L intact OPN, peptide NT 109 to 153 or peptide CT 154 to 198 after 120 mins OGD. A total of 24 h after OGD, 20μL of MTT (5 mg/mL) was added directly to the media in each well for 1 h. The medium was removed and 100 μL of dimethyl sulfoxide added to each well. The plates were then read at 550 nm and compared with plates that had not undergone OGD (non-OGD control).
Western Blotting
Western blotting was performed as described previously (Meller et al, 2005). Protein was denatured in a gel-loading buffer at 100°C for 5 mins and then loaded onto a 12% SDS-polyacrylamide gel. Protein was then transferred to a polyvinylidene difluoride membrane and incubated with primary goat anti-mouse OPN antibody (Sigma) at 4°C overnight. Membranes were rinsed and incubated with donkey anti-goat IgG conjugated to horseradish peroxidase followed by chemiluminescence detection (NEN Life Science Products, Boston, MA, USA).
Mice
C57Bl/6mice (8- to 12-week-old males and females) were obtained from Jackson Laboratory (Bar Harbor, ME, USA). All procedures met NIH guidelines with the approval of the Oregon Health & Science University Institutional Animal Care and Use Committee. The total number of mice used for this study was 260 (248 males and 12 females).
Surgery
Focal cerebral ischemia was induced by middle cerebral artery occlusion (MCAO) as described previously (Clark et al, 1997). Briefly, mice were anesthetized by isoflurane inhalation (4%/L O2) and maintained with 1.5%/L O2. The middle cerebral artery was occluded by threading a silicone-coated 8-0 monofilament nylon surgical suture through the external carotid to the internal carotid, blocking its bifurcation into the MCA and anterior cerebral artery. The filament was kept in the vessel lumen for 75 or 60 mins while the mice were maintained under anesthesia. The filament was then removed, thereby restoring blood flow. Cerebral blood flow was monitored throughout the surgery by laser-Doppler flowmetry (Periflow 5000; Perimed, Sweden) to confirm that during the surgery, blood flow in the MCA was less than 10% of baseline. Laser-Doppler monitoring ensures a rate of infarct success of >95%. Furthermore, the survival rate of this surgery in our hands is >90%. Body temperature was monitored throughout the surgery using a rectal thermometer (Hi-Lo Temp 8200 Patient Temperature Monitor; Sensortek Inc., Lake forest, CA, USA). After surgery, mice were kept on a thermal barrier pad with free access to soft food and were killed after 24 or 72 h. Core temperature and blood glucose levels were monitored after OPN treatment in the presence and absence of MCAO and no differences were noted between animals treated with OPN and those administered vehicle (data not shown). Mice administered intracerebroventricular injections were exposed to MCAO for 75 mins, whereas mice undergoing intranasal administrations were exposed to MCAO for 60 mins. The extended duration of MCAO used in the setting of intracerebroventricular injections (75 mins) was used because the procedure of intracerebroventricular injection itself confers a modest, but measurable, amount of neuroprotection perhaps because of mild spreading depression, which occurs on insertion of the intracerebroventricular cannula (Strong and Dardis, 2005). The procedure of intranasal administration does not induce neuroprotection; thus, MCAO duration of 60 mins was used in experiments that required this route of drug delivery.
Intracerebral Ventricular Injections
Vehicle (artificial cerebrospinal fluid (aCSF)) or equimolar amounts of OPN or thrombin-treated OPN were injected into the left lateral ventricle (coordinates: 0 mm bregma, 1 mm lateral, and 2.5 mm ventral) using a 27-gauge needle. Artificial CSF (1 μL), thrombin-treated OPN (0.1 μg/μL), or intact OPN (0.1 μg/μL) were injected immediately after the MCAO surgery.
Infarct Measurement
Mice were deeply anesthetized with isoflurane and then perfused with heparinized saline (2 U/mL) via the ascending aorta at a flow rate of 9 mL/min. Brains were rapidly removed, placed on a tissue slicer (Stoelting, Wood Dale, IL, USA), and covered with soft agarose. The olfactory bulb and cerebellum were removed and discarded. The remaining brain was sectioned into 1 mm slices beginning from the rostral end. To visualize the region of infarction, sections were placed in 1.5% 2,3,5-triphenyltetrazolium chloride in 0.9% PBS and stained for 15 mins at 37°C. As 2,3,5-triphenyltetrazolium chloride staining is a measure of metabolic function, cells that fail to take up this vital dye are viewed as nonviable. Accordingly, unstained brain tissue is measured and considered the region of infarct. After staining, the sections were transferred to 10% formalin. Images of the sections were scanned, and the area of the infarct and the ipsilateral hemisphere were measured using NIH image 1.62 by a technician blinded to the treatment group. The measurements were multiplied by the thickness of the section (1 mm) and then summed over the entire brain to yield volume measurements. Ischemic damage data are presented as percent injury of the ipsilateral hemisphere, ((infarct volume)/(ipsilateral hemisphere volume) × 100), rather than absolute volume. This equation corrects for hemispheric edema, which is found in both infarcted and healthy tissue within the ipsilateral hemisphere (Lin et al, 1992). This direct infarct assessment method is appropriate for ‘percent’ infarct volume measurements as presented here, whereas the indirect method (Swanson et al, 1990) is used for ‘absolute’ measurements of infarct size (i.e., in cubic millimeter).
Neurologic Assessment
Animals were scored for focal neurologic deficits 24 h after MCAO. Scoring was based on physical appearance and behavior using a scale previously developed to provide a precise assessment of focal neurologic damage (Hill et al, 1999). The mice were scored in each of the following seven categories: body symmetry, gait, climbing, circling behavior, forelimb symmetry, compulsory circling, and sensory response. Each mouse was scored from 0 (no deficit) to 4 (severely affected) in each category.
Intranasal Administration of Osteopontin
Intranasal administration was performed as follows: mice were placed on their backs and administered either aCSF, OPN, thrombin-treated OPN, or OPN peptides, in nose drops (5 μL/drop) over a period of 20 mins, alternating drops every 2 to 5 mins between the left and right nares. For studies of neuroprotection, the total volume delivered was 50 μL. Nasal administration of thrombin-cleaved OPN (5 μg) was started 10 mins after initiating MCAO. Osteopontin peptides were delivered intranasally either 10 mins after initiating MCAO or at 1, 3, or 5 h after MCAO. For all experiments except the dose—response experiment, the dose of each peptide administered was equimolar to 5 μg of thrombin-cleaved OPN (Table 1). For the dose—response experiment of NT 134 to 153, the peptide was delivered 1 h after initiating MCAO at a dose 10-fold higher and 10-fold lower than the previously identified protective dose (350 ng).
Dose of each peptide that is equimolar to 50 μg of thrombin-cleaved OPN
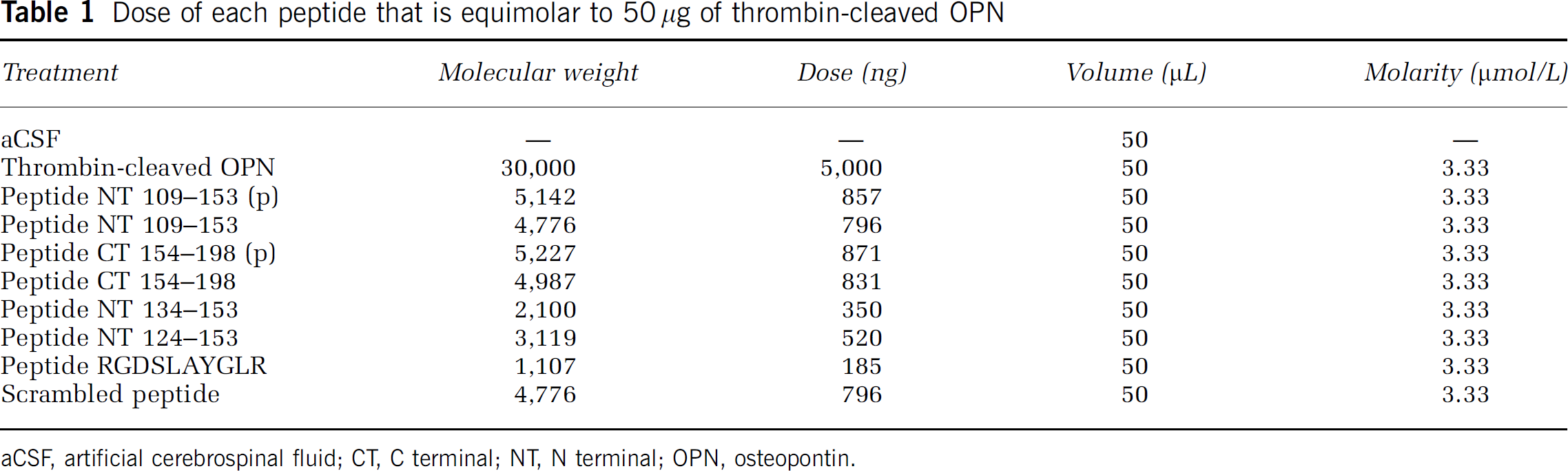
aCSF, artificial cerebrospinal fluid; CT, C terminal; NT, N terminal; OPN, osteopontin.
Detection of Osteopontin in the Brain after Nasal Administration
Immunofluorescent detection of OPN in the brain was performed on mice administered 2 μg (20 μL) of OPN or aCSF. Mice were killed 2 h after administration (n=3 per group). Brains were sliced on a cryostat to a section thickness of 20 μm. The OPN antibody used was MPIIIB10 (Developmental Studies Hybridoma Bank, Iowa City, IA, USA) and the secondary antibody used was mouse anti-mouse IgG conjugated to Cy 3. Tissue was mounted in Vectashield mounting medium containing DAPI.
Enzyme-linked immunosorbent assay detection of OPN in the brain was performed on mice that received 5 μg (50 μL) of OPN or aCSF. The CSF was tapped from mice killed at 30 mins, 1 and 2 h after administration, by puncturing the cisterna magna with a 27-gauge needle connected to a syringe (n=3 per group). The OPN concentration was calculated using an enzyme-linked immunosorbent assay kit for mouse (Assay Designs TiterZyme EIA kit for mouse OPN, Ann Arbor, MI, USA) according to the manufacturer's instructions. This kit can only detect full-length OPN; thus, only the concentration of intact OPN could be evaluated in the brain after intranasal administration.
Statistical Analysis
Data are shown as means±s.e.m. of n determinations unless otherwise indicated. Data from cell viability assays and in vivo stroke experiments were analyzed by one-way ANOVA followed by Bonferroni's multiple comparison test, using Graphpad Prism version 4.0 (Graphpad software, San Diego, CA, USA).
Results
Treatment of Osteopontin with Thrombin
We previously reported that recombinant mouse OPN confers protection against ischemic injury via ligation of integrin receptors and activation of phosphoinositide-3 kinase and mitogen-activated protein kinase pathways (Meller et al, 2005). Here, we sought to determine whether the neuroprotective effect of OPN could be improved by thrombin cleavage. Recombinant mouse OPN is produced as a mixture of glycosylated and nonglycosylated OPN proteins. We found that incubation with thrombin resulted in efficient cleavage of glycosylated OPN, but not nonglycosylated OPN (Figure 2A). The nonglycosylated form of OPN remained resistant to cleavage despite incubation in thrombin for 72 h at 37°C. This suggests that glycosylation may be required for recognition and/or cleavage of OPN by thrombin.
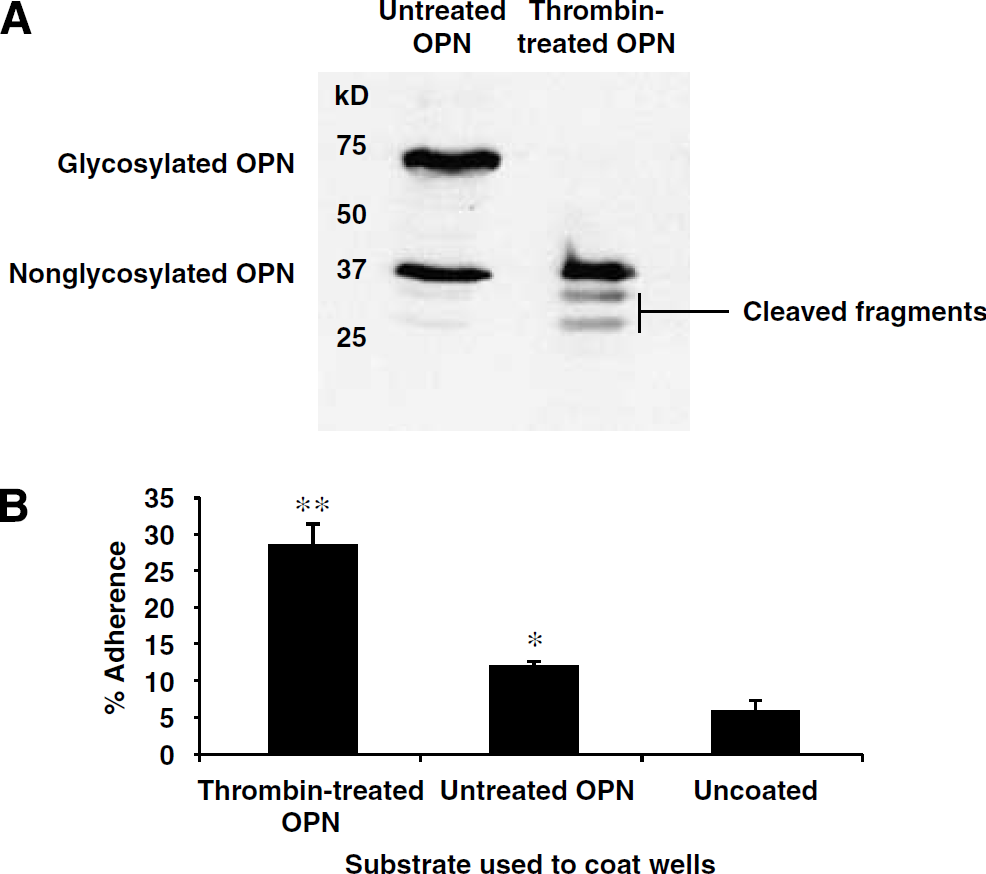
(
Thrombin Treatment Improves Integrin-Binding Ability
Treatment of OPN with thrombin generated the following products: intact nonglycosylated OPN and two peptide fragments representing the C- and N-terminal fragments of glycosylated OPN. Thrombin cleavage led to a substantial enrichment of the cleaved fragments of OPN (Figure 2A). Treatment with thrombin improves the ability of OPN to ligate integrin receptors (Senger et al, 1994); thus, we tested whether thrombin treatment of mouse OPN improved cell adhesion using HEK 293 cells. We found that cells plated on thrombin-treated OPN displayed an improved ability to adhere to their substrate compared with cells plated on untreated, intact OPN (Figure 2B).
Thrombin Treatment of Osteopontin Improves Neuroprotection In Vitro
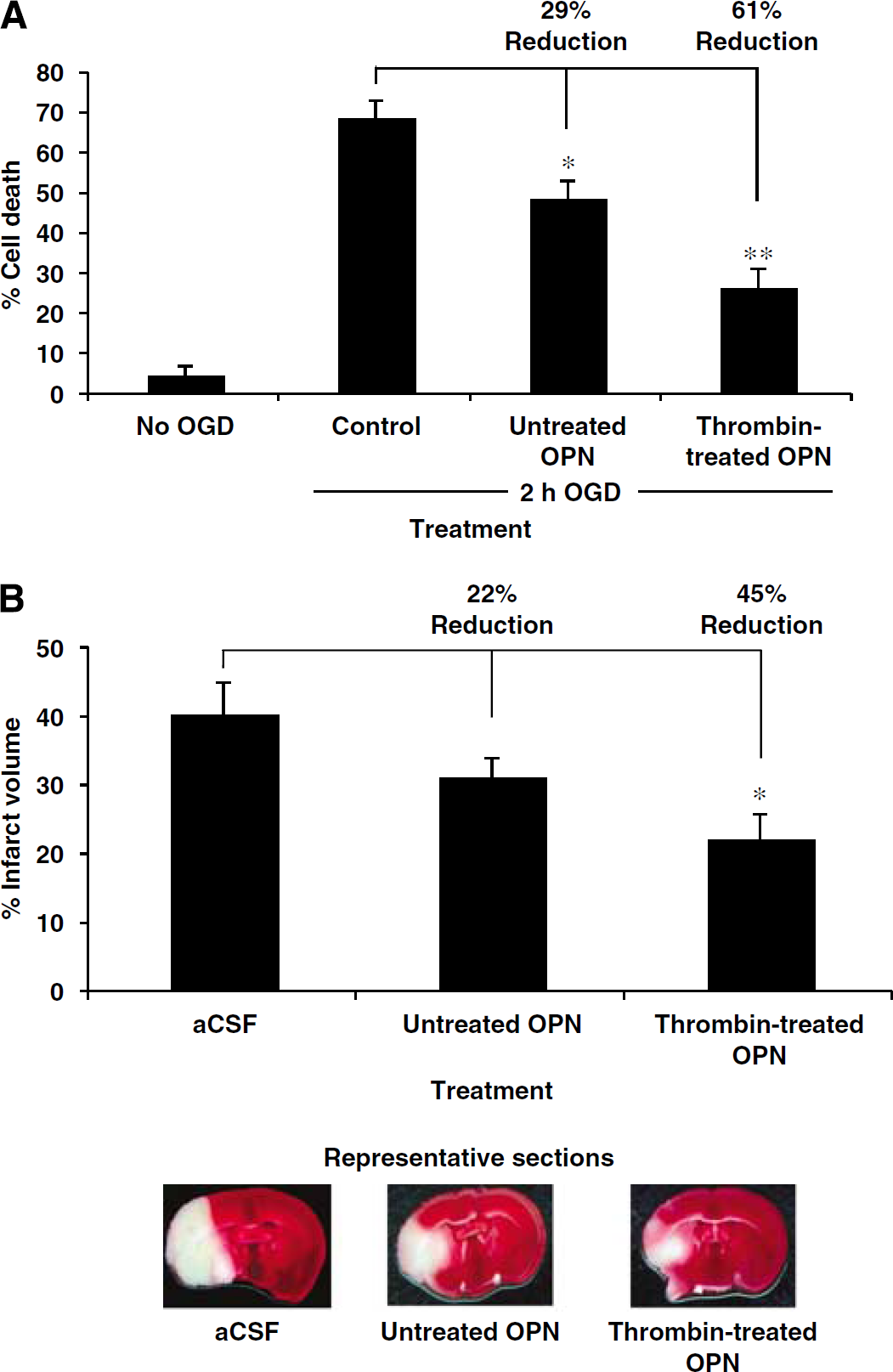
We previously showed that neuroprotection induced by OPN occurs via ligation of integrin receptors and depends on RGD interactions (Meller et al, 2005). Thus, we tested whether thrombin cleavage of OPN, which occurs near the RGD site, improves the neuroprotective capacity of OPN. Primary rat neuronal cultures were exposed to OGD followed by incubation with untreated or thrombin-treated OPN (5 nmol/L) for 24 h after OGD. Intact OPN conferred significant protection against OGD-induced cell death, as we expected based on our previously published results (Meller et al, 2005). Importantly, however, thrombin-treated OPN provided substantially greater protection against OGD-induced cell death compared with untreated OPN (61% versus 29% reduction in cell death; Figure 3A).
Thrombin Treatment Improves Osteopontin-Mediated Neuroprotection In Vivo
Thrombin-treated OPN, untreated OPN, or aCSF was administered intracerebroventricularly immediately after MCAO (75 mins) and the degree of neuroprotection was compared 24 h later. Thrombin-treated OPN reduced infarct volume twofold more than untreated OPN (45% versus 22% reduction) relative to vehicle-treated animals (Figure 3B). Untreated OPN provided protection against ischemic injury, as we had found previously; however, in this experiment, the effect did not reach statistical significance.
Intranasal Administration of Thrombin-Treated Osteopontin Protects Against Stroke Injury
The therapeutic potential of a treatment for brain injury can be improved by intranasal delivery to the CNS. Here, we used immunofluorescence to test whether OPN reaches the brain after intranasal administration. Within 2 h after intranasal administration of 2 μg OPN, the presence of OPN was detected in brain by immunostaining. The pattern of staining showed high OPN immunoreactivity in the striatum and cortex of the brain (Figure 4A). This pattern of staining shows that intranasal administration can deliver OPN to the territory rendered ischemic by MCAO. To quantify the amount of OPN that reaches the brain after intranasal delivery, CSF was sampled from mice treated with 5 μg of OPN or aCSF at 30 mins, 1 h, and 2 h after administration. We measured OPN levels in the CSF rather than actual brain tissue because our aim in administering OPN was to increase the extracellular concentration of the protein. The OPN measurements of brain tissue would have been confounded by the inclusion of endogenous intracellular OPN. The concentration of OPN increased by 50%, 2 h after OPN administration (Figure 4B).
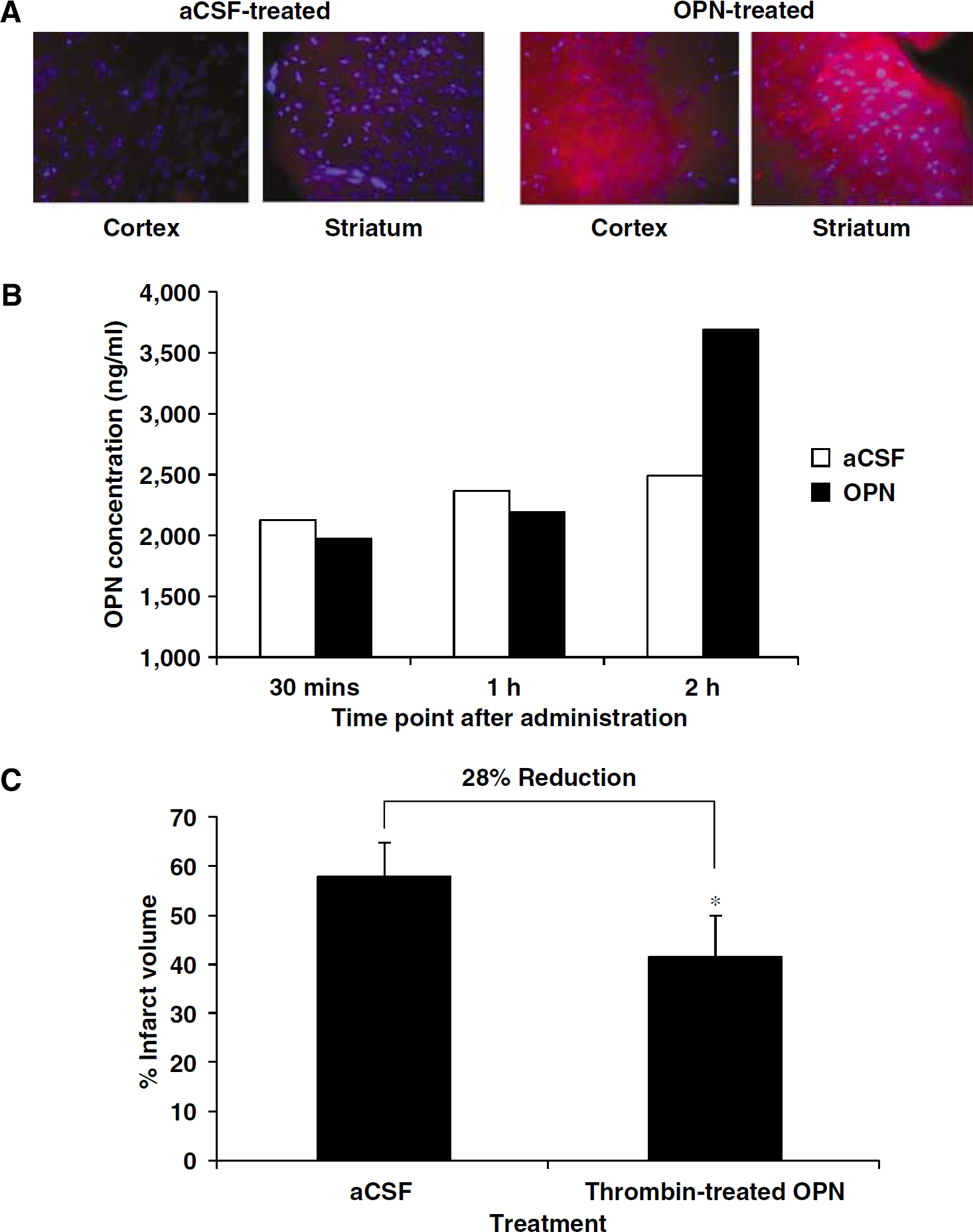
Osteopontin (OPN) delivery to the brain via intranasal administration. (
To allow adequate delivery and accumulation of OPN in the brain at the time of stroke injury, we administered thrombin-treated OPN (5 μg) intranasally 10 mins after the initiation of 60 mins MCAO. Although the majority of infarct damage is evident within 24 h of MCAO, the infarct evolves further over the next several days. Hence, infarct measurements were made 72 h after MCAO to provide a more accurate picture of neuroprotection and to distinguish whether treatment is truly neuroprotective or merely delays damage (Pantano et al, 1999). We found that intranasal administration of thrombin-treated OPN significantly reduced infarct volume by 28% compared with vehicle-treated mice (Figure 4C).
Peptides Based on the N- and C-Terminal Fragments of Thrombin-Cleaved OPN are Neuroprotective
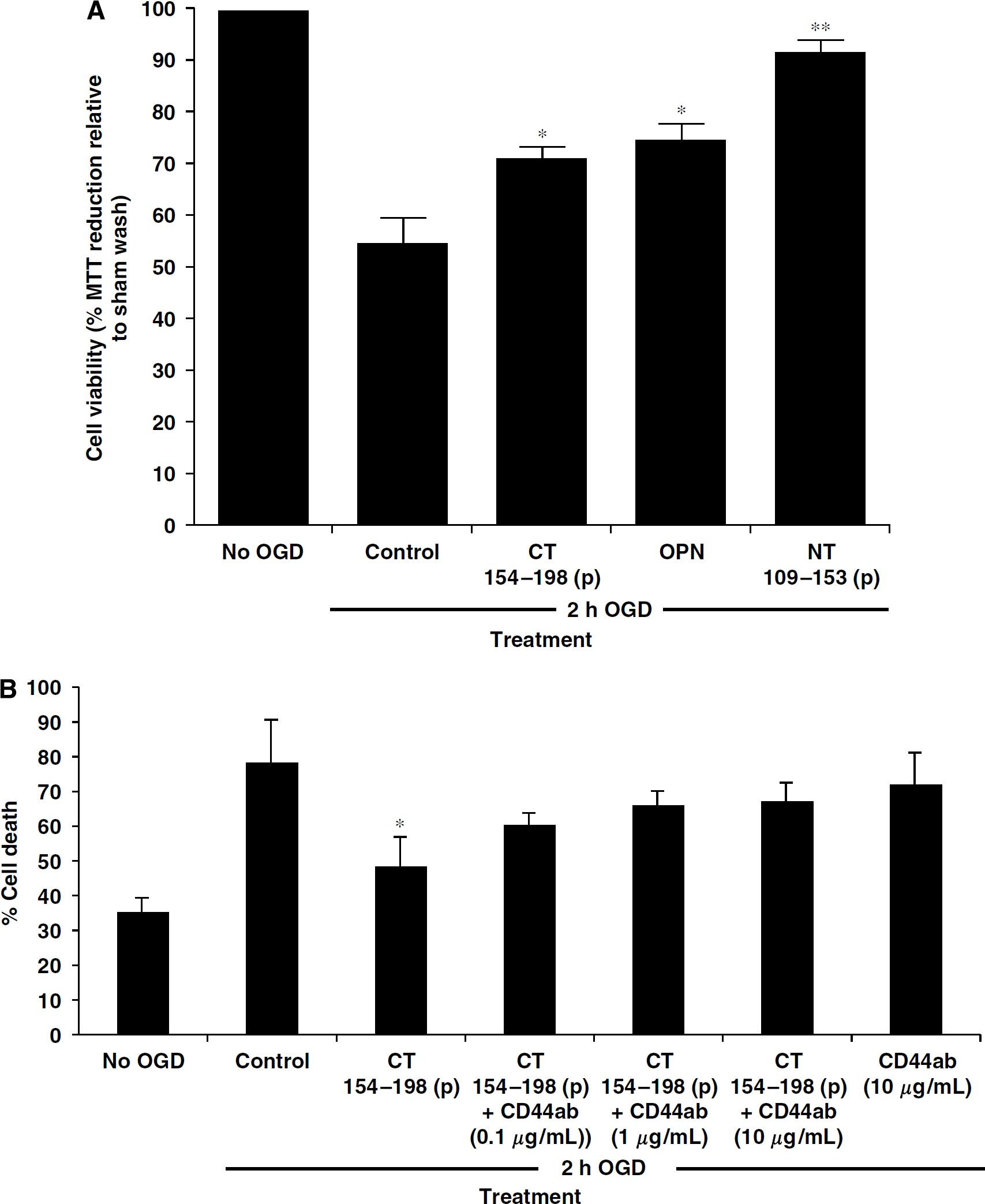
We next sought to test whether one or both of the peptide fragments generated from thrombin cleavage of OPN were neuroprotective. Two synthetic 45-amino-acid peptides corresponding to the sequence immediately N-terminal and C-terminal to the thrombin cleavage site of OPN were evaluated for neuroprotective capacity. These peptides were generated with phosphorylated residues at sites reported to be phosphorylated in native OPN (Sorensen et al, 1999). NT 109 to 153 (p), the N-terminal peptide corresponding to amino acids 109 to 153 of native OPN, which contains the integrin-binding sequences RGD and SLAYGLR, conferred greater neuroprotection than intact OPN in our in vitro model of stroke when compared at an equivalent molar dose (5 nmol/L) (Figure 5A). Unexpectedly, we also found pronounced neuroprotection conferred by the C-terminal peptide, CT 154 to 198 (p), which is based on the sequence of the C-terminal fragment of OPN. This result was surprising because we previously found that OPN mediates neuroprotection by interaction with integrin receptors, a property associated with the region of OPN N-terminal to the thrombin cleavage site. Based on previous reports that the C-terminal half of OPN contains a CD44-binding motif, we tested whether the neuroprotective effect of this peptide could be abrogated by a CD44-neutralizing antibody. Cultures were treated with 5 nmol/L CT 154 to 198 (p) in the presence of 0.1 to 10 μg/mL of a CD44-neutralizing antibody. The neuroprotective effect of CT 154 to 198 (p) was diminished in the presence of the antibody, suggesting that this peptide protects, at least in part, via CD44 ligation (Figure 5B).
Based on our finding that peptide NT 109 to 153 (p) and peptide CT 154 to 198 (p) conferred neuroprotection in modeled ischemia in vitro, we tested each peptide for its ability to attenuate stroke injury in vivo. We found that each peptide when delivered intranasally at a dose equimolar to the protective dose of thrombin-treated OPN reduced ischemic damage. NT 109 to 153 (p) and CT 154 to 198 (p) reduced infarct volume by 36% and 34%, respectively (Figure 6). For this experiment, the peptides were administered 1 h after MCAO, validating that there is a time window of at least 1 h available for treatment with these peptides.
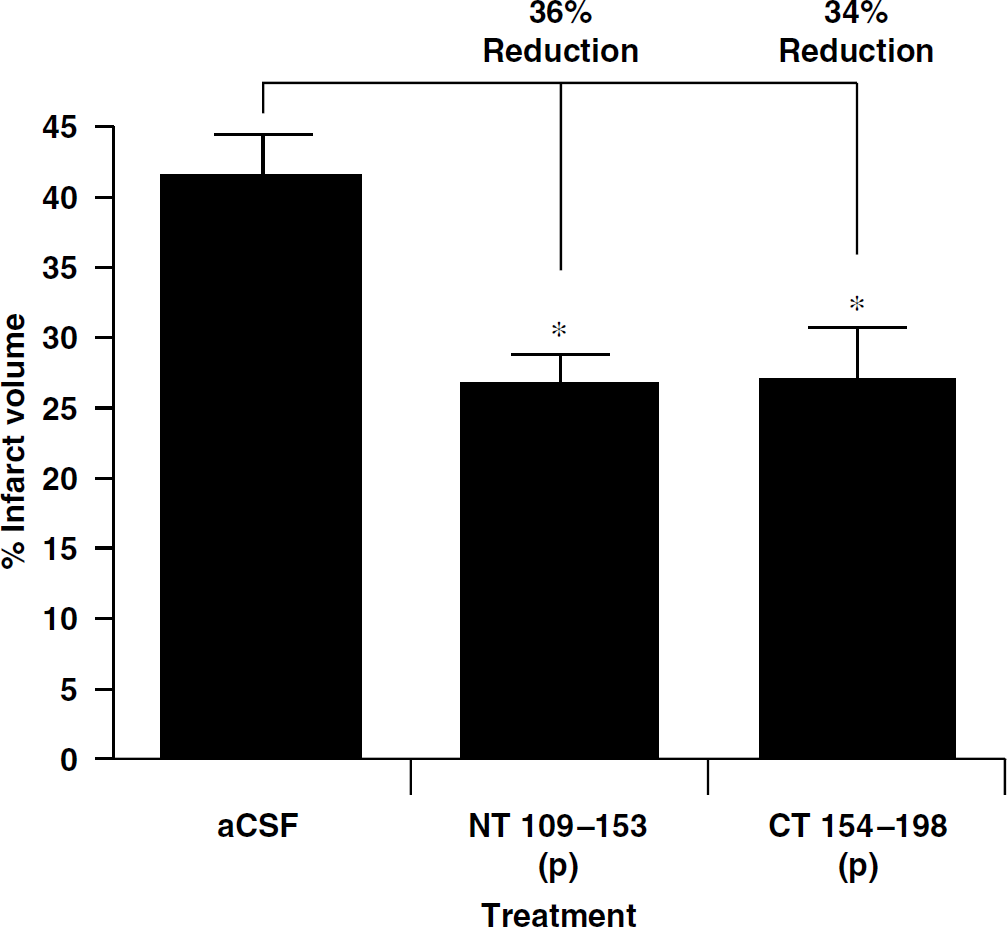
Intranasal administration of peptides NT 109 to 153 (p) or CT 154 to 198 (p) reduces infarct volume after middle cerebral artery occlusion (MCAO). One hour after MCAO, peptides were administered intranasally at a dose equimolar to 5 μg of OPN. Mice were killed 24 h later. ∗P<0.05 compared with artificial cerebrospinal fluid administration (n=8 per group).
The C-Terminal but not N-Terminal Peptide must be Phosphorylated for Neuroprotection
To determine whether peptides NT 109 to 153 (p) and CT 154 to 198 (p) require phosphorylation for their neuroprotective activity, we synthesized and tested the nonphosphorylated versions of each peptide (peptides NT 109 to 153 and CT 154 to 198; Figure 7A). These peptides were administered intranasally 10 mins after initiating MCAO at a concentration equimolar to a dose of 5 μg of thrombin-treated OPN. The nonphosphorylated C-terminal peptide failed to provide neuroprotection, whereas the nonphosphorylated N-terminal peptide retained neuroprotective capability. A scrambled 45-amino-acid peptide based on the sequence of NT 109 to 153 also failed to provide neuroprotection (Figure 7B).
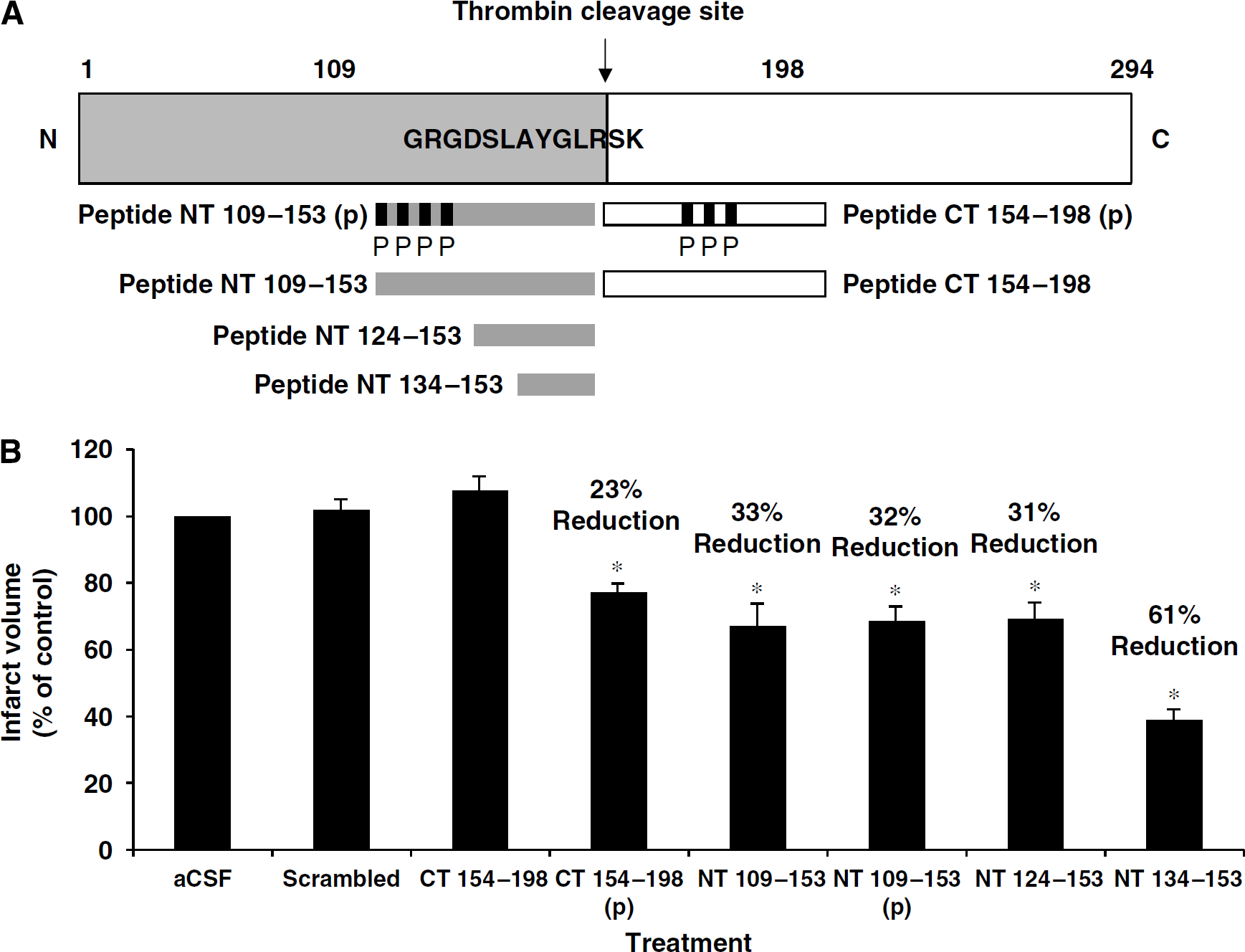
Nonphosphorylated truncated peptides of NT 109 to 153 (p) confer robust neuroprotection. (
Peptide NT 109 to 153 (p) contains the integrin-binding motif RGDSLAYGLR at its distal end, whereas the phosphorylated residues are located proximal to the N terminus of the peptide (Figure 7A). In light of our finding that this peptide does not require phosphorylation to confer neuroprotection, we tested additional N-terminal OPN peptides that were further truncated at the N terminus. The 30-amino-acid peptide NT 124 to 153 and the 20-amino-acid peptide NT 134 to 153 were administered intranasally 10 mins after initiating MCAO and mice were killed 24 h later. Each of these peptides conferred significant protection against ischemic injury. NT 124 to 153 reduced infarct volume by 31% and NT 134 to 153 reduced infarct volume by 61% (Figure 7B). These data suggest that the neuroprotective sequence of the N-terminal fragment of OPN lies between residues 134 and 153 and provides a relatively short sequence for future drug design. To determine whether the reduction in infarct volume seen with NT 134 to 153 was associated with improved neurologic function, 12 more mice were administered either 350 ng NT 134 to 153 or aCSF during MCAO and scored for focal neurologic deficit 24 h after stroke. Mice that received the peptide showed less neurologic impairment compared with control mice (Figure 8).
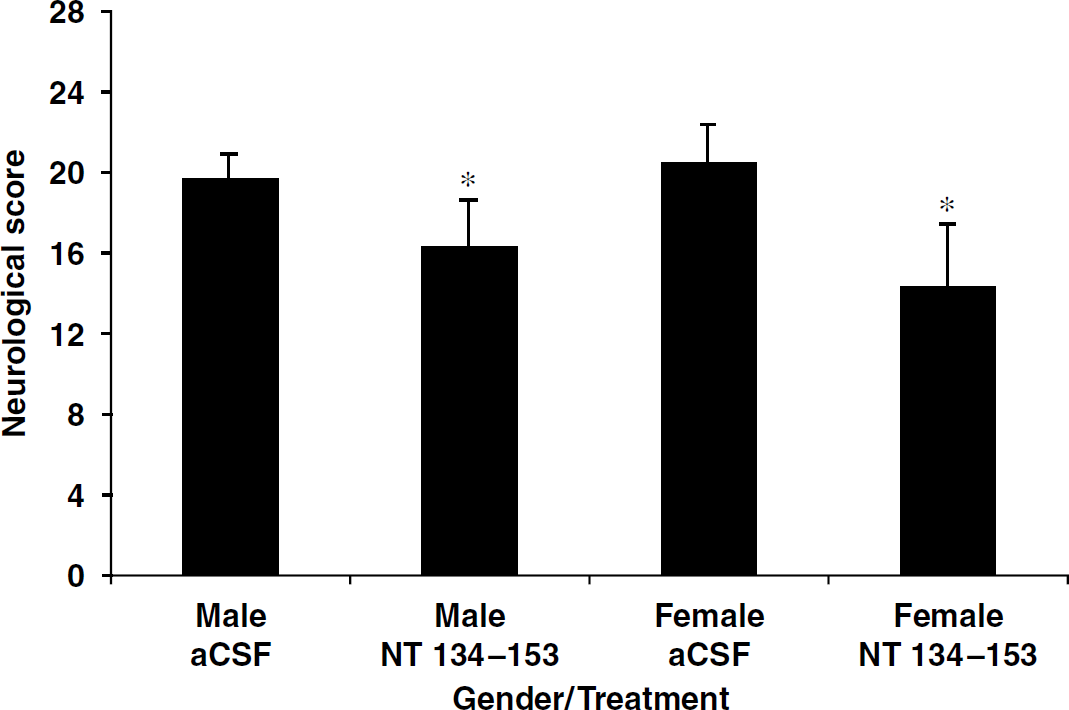
Neurologic scoring in males and females after treatment with NT 134 to 153. Mice were killed 24 h after middle cerebral artery occlusion. ∗P<0.05 compared with artificial cerebrospinal fluid administration (n=6 per group). Data shown are means±s.d. Higher scores indicate greater neurologic impairment.
The Effect of NT 134 to 153 in Females
To further explore the clinical potential of peptide NT 134 to 153, we tested whether the peptide is neuroprotective in females. The peptide was delivered intranasally during MCAO at the dose that proved neuroprotective in males. In females, the peptide reduced infarct volume by 25% (data not shown) and resulted in a significant improvement in neurologic function compared with females treated with aCSF (Figure 8).
Dose—Response and Time Window of NT 134 to 153
To investigate the relationship of dose and time on the efficacy of peptide NT 134 to 153, we tested doses that were 10-fold higher and lower than the previously identified protective dose at 1, 3, and 5 h after MCAO. When delivered intranasally 1 h after MCAO, NT 134 to 153 conferred a similar amount of protection at 3,500 and 350 ng, but was no longer neuroprotective at 35 ng (Figure 9A). At a dose of 350 ng, NT 134 to 153 conferred a significant amount of protection at 3 h, but not when delivered at the later time point of 5 h after MCAO (Figure 9B). This indicates that there is a time window of at least 3 h available for treatment with this peptide.
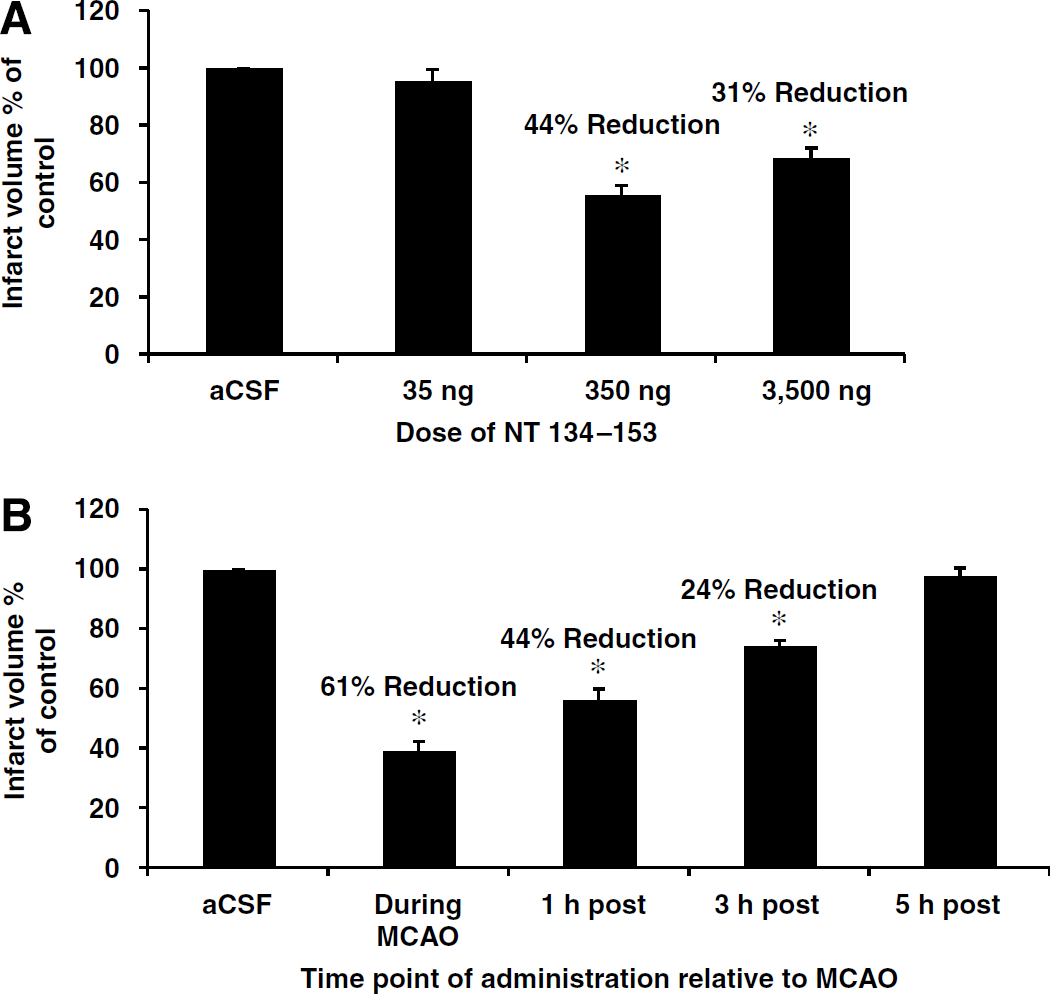
Time window and dose—response of NT 134 to 153. (
The 10-Amino-Acid Fragment, RGDSLAYGLR, Found in NT 134 to 153 Confers Neuroprotection
The integrin-binding sequence of peptide NT 134 to 153 consists of the consecutive residues ‘RGDSLAYGLR’. To determine whether this region of peptide NT 134 to 153 is sufficient for neuroprotection, we synthesized a decamer peptide consisting solely of the sequence ‘RGDSLAYGLR’. Administration of ‘RGDSLAYGLR’ during MCAO conferred an equivalent amount of neuroprotection as peptide NT 134 to 153 (Figure 10). This result indicates that the decamer peptide ‘RGDSLAYGLR’ is a promising candidate for the design of a peptidomimetic for use in the treatment of ischemic brain injury.
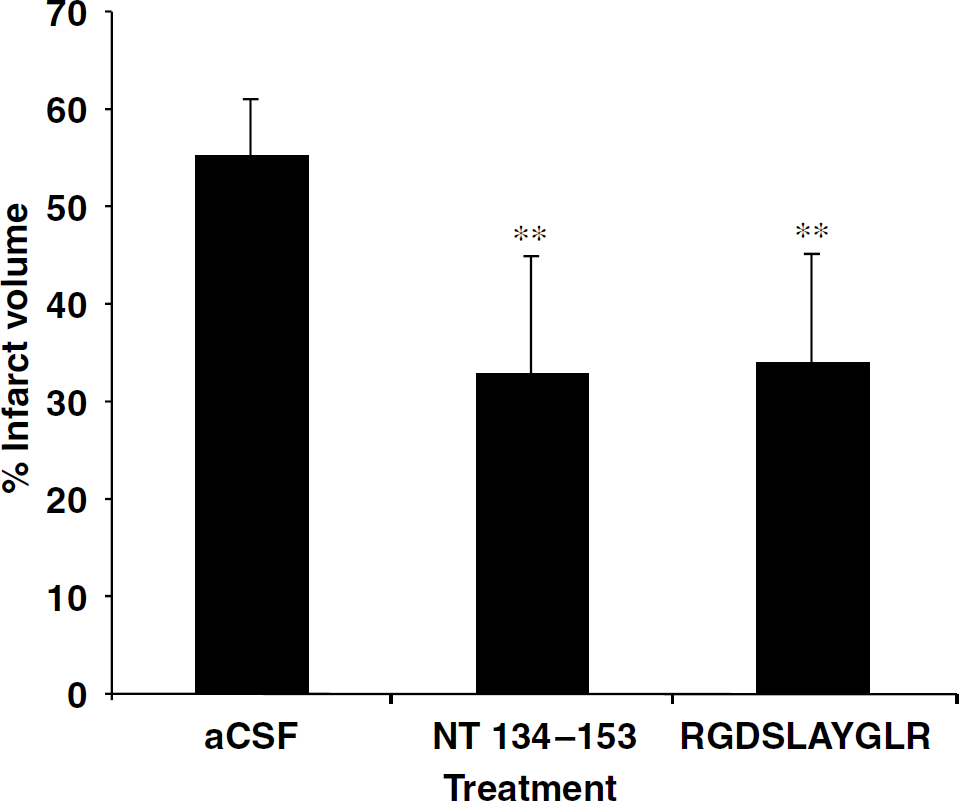
NT 134 to 153 can be truncated to the decamer peptide ‘RGDSLAYGLR’. During middle cerebral artery occlusion, peptides were administered intranasally at a dose equimolar to 5 μg of osteopontin. Mice were killed 24 h later. ∗∗P<0.001 compared with artificial cerebrospinal fluid (aCSF) administration (n=14 aCSF group, n=9 ‘NT 134 to 153’ group, n=6 ‘RGDSLAYGLR’ group). Data shown are means±s.d.
Discussion
Intact and thrombin-cleaved OPN have distinct integrin-binding properties (Senger et al, 1994). Here, we tested the hypothesis that thrombin treatment of OPN would enhance its neuroprotective features. We found that glycosylated, but not nonglycosylated, recombinant mouse OPN is cleaved by thrombin. This suggests that glycosylation of OPN plays an important role in thrombin recognition and proteolysis of OPN. The active site of thrombin is regulated by two exosites, one of which, exosite II, can bind to saccharide chains (Carter et al, 2005). Oligosaccharides closely packed together on the polypeptide chain of OPN may constitute a saccharide patch that enhances recognition by thrombin or regulates thrombin function (Varki, 1993). Recent evidence supports this hypothesis wherein OPN derived from human milk consists of O-linked glycosylation, with all of the sites of glycosylation lying in close proximity to the thrombin cleavage site (Sorensen et al, 1995). Interestingly, the synthetic peptides used in this study were not glycosylated, which indicates that OPN confers neuronal survival in a manner independent of glycosylation.
Thrombin-treated OPN was significantly more effective than untreated OPN in promoting HEK 293 cell adherence, which suggests that thrombin treatment improves the ability of OPN to bind integrin receptors. We reasoned that improved binding of OPN may enhance neuroprotection. The studies described here show that compared with intact OPN, thrombin-cleaved OPN provides twofold greater neuroprotection in ischemic stroke models.
The therapeutic potential of OPN treatment in stroke may be improved with intranasal administration—a means of delivery that targets proteins to the brain (Born et al, 2002). Intranasal administration has several advantages over other methods of drug delivery to the brain, as it is noninvasive, simple to perform, and targets the brain >100-fold more than the circulation (Thorne and Frey, 2001). A recent study showed a reduction in stroke volume and improved behavioral scores for rats that were administered insulin-like growth factor-I intranasally after MCAO (Liu et al, 1992). An additional study has shown that treatment of stroke can be successful using the intranasal delivery of the antioxidant myricetin (Liu et al, 1992). Our studies add to the growing list of neuroprotectants that can be delivered intranasally, as we show that when 5 μg of OPN is administered via the intranasal route, OPN levels in the brain are increased by 50%.
In view of the improved neuroprotection conferred by thrombin-treated OPN, we examined the neuroprotective capacity of two synthetic 45-amino-acid peptides representing the N- and C-terminal halves of thrombin-cleaved OPN. The use of synthetic peptides allowed us to test fragments of OPN individually and without the contaminating presence of the intact nonglycosylated molecule found in recombinant OPN preparations.
The N-terminal peptide, NT 109 to 153 (p), which contains the RGD motif, conferred marked neuroprotection in the setting of ischemia. Moreover, when this peptide was tested in vitro, it conferred significantly more neuroprotection than intact OPN when tested at an equimolar dose. This is consistent with our hypothesis that thrombin treatment of OPN improves the ability of the RGDSLAYGLR sequence to bind to integrin receptors. However, it is somewhat surprising that the C-terminal peptide, CT 154 to 198 (p), which lacks an RGD motif, also displayed neuroprotective activity in vitro and in vivo. This means that the improved neuroprotective capacity of thrombin-treated OPN may not be due solely to improved integrin-binding ability. It is possible that increased potency may be because of the mobilization of the C-terminal end of OPN after thrombin cleavage, thereby permitting the C-terminal fragment to become untethered from the N-terminal fragment and allow more receptors to be bound by thrombin-cleaved OPN. For example, in addition to binding integrin receptors, OPN is a ligand for CD44, a receptor that mediates a diverse array of functions, including cell migration, leukocyte activation, cell communication, and cell survival (Larkin et al, 2006). CD44 has an antiapoptotic effect in lymphocytes and the loss of CD44 promotes colon carcinoma apoptosis (Ayroldi et al, 1995; Bates et al, 2001). As the C-terminal fragment of OPN contains a CD44-binding site, such cell survival properties may underlie the neuroprotective role of peptide CT 154 to 198 (p). In support of this hypothesis, the neuroprotective effect of CT 154 to 198 (p) was reduced when cells were treated with CT 154 to 198 (p) in the presence of a CD44-neutralizing antibody.
To test whether peptides CT 154 to 198 (p) and NT 109 to 153 (p) require phosphorylation for their neuroprotective activity, we tested the nonphosphorylated versions of each of these peptides. We found that CT 154 to 198 did not confer neuroprotection in the absence of phosphorylated residues at sites threonine 170, serine 176, and serine 180.
Peptide NT 109 to 153 was neuroprotective in the absence of phosphorylation. This indicates that the N-terminal fragment of OPN does not require phosphorylation to enhance neuronal survival. This is consistent with a previous report that mapped the structural requirements of the N-terminal half of OPN necessary for binding to integrins αvβ6, αvβ3, αvβ5, α5β1, and α9β and found that phosphorylation is not a prerequisite (Yokosaki et al, 2005).
We further tested whether peptide NT 109 to 153 could be truncated via removal of residues from the N terminus and retain neuroprotective capability. We tested a 30- and a 20-amino-acid version of this peptide, each containing an intact RGDSLAYGLR sequence, for the ability to protect the brain from ischemic injury. We found that intranasal delivery of either peptide was neuroprotective. Interestingly, peptide NT 134 to 153, which is the shorter of the two, displayed greater neuroprotection. This result may reflect that the smaller molecular size of NT 134 to 153 results in superior delivery because molecular size correlates inversely with delivery to the brain via the intranasal route (Fisher et al, 1992; Sakane et al, 1995).
We selected peptide NT 134 to 153 for additional preclinical testing based on its greater efficacy when compared at an equimolar concentration with the other OPN peptides. We found that this peptide is neuroprotective in females and that reduced infarct volume is associated with attenuated neurologic impairment. A dose—response experiment was performed to determine the potency of NT 134 to 153 and whether delivering a higher dose increases efficacy. A 10-fold higher dose was not more neuroprotective than the previously identified protective dose and a 10-fold lower dose failed to confer neuroprotection. We also investigated the time window available for administration and found that NT 134 to 153 confers neuroprotection when delivered at 1 and 3 h after MCAO, but not at 5 h after MCAO. This experiment also revealed that NT 134 to 153 confers the greatest amount of neuroprotection when delivered during ischemia. This offers promise to patients scheduled for coronary artery bypass surgery, of which approximately 50% suffer permanent cognitive decline from intraoperative emboli (Newman et al, 2001).
To further isolate the region of peptide NT 134 to 153 required for neuroprotection, we tested a decamer peptide consisting solely of the sequence ‘RGDSLAYGLR’. We found that this shortened version of NT 134 to 153 conferred an equivalent amount of neuroprotection, providing a relatively short sequence for the design of a peptidomimetic.
In summary, thrombin-treated OPN is superior to the intact molecule in conferring neuroprotection. Moreover, synthetic peptides based on the N- or C-terminal portions of OPN confer neuroprotection when administered alone. It is noteworthy that these peptides protect against stroke injury when delivered intranasally—a feature that offers substantial hope for their future therapeutic application. We anticipate that additional drugs based on the sequence of OPN may emerge as important components of future stroke therapy.