
Abstract
Inhibition of the canonical Wnt pathway has been implicated in the pathophysiology of neuronal death. Here, we report that the secreted Wnt antagonist, Dickkopf-1 (Dkk-1) is rapidly induced in neurons after induction of focal brain ischemia. In rats undergoing transient focal ischemia in response to brain infusion of endothelin-1, Dkk-1 was induced in neurons of the ischemic core and the penumbra region. Induction of Dkk-1 was associated with a reduced expression of β-catenin (a downstream signaling molecule of the canonical Wnt pathway), and was not observed in neurons expressing the protective protein, heat shock protein-70. Treatment with lithium ions, which, inter alia, rescue the canonical Wnt pathway, was highly protective against ischemic damage. Dkk-1 was also induced in cortical neurons of mice undergoing permanent middle cerebral artery (MCA) occlusion. This model allowed us to compare wild-type mice with doubleridge mice, which are characterized by a reduced expression of Dkk-1. Doubleridge mice showed an attenuated reduction of β-catenin and a reduced infarct volume in response to MCA occlusion, providing a direct demonstration that Dkk-1 contributes to the pathophysiology of ischemic neuronal damage. These data rise the interesting possibility that Dkk-1 antagonists or drugs that rescue the Wnt pathway might be neuroprotective in stroke.
Introduction
Reperfusion induced by recombinant tissue plasminogen activator is the mainstay treatment for thromboembolic stroke (The National Institute of Neurological Disorders and Stroke rt-PA Stroke Study Group, 1995). However, the narrow time window (3–6 h after stroke) and the risk of hemorrhagic complications limit the use of tissue plasminogen activator to a minority of patients with stroke (Heuschmann et al, 2004). In addition, animal studies suggest that tissue plasminogen activator paradoxically enhances neuronal death by cleaving and activating N-methyl-
A growing body of evidence shows that inhibition of the canonical Wnt pathway contributes to the pathophysiology of neuronal damage in model of acute and chronic neurodegenerative disorders (De Ferrari and Inestrosa, 2000; Garrido et al, 2002; Inestrosa et al, 2002; Caricasole et al, 2003, 2004; De Ferrari et al, 2003; Cappuccio et al, 2005; Scali et al,2006; Busceti et al, 2007). Wnts are members of a family of secreted glycoproteins that regulate neuronal homeostasis during development, as well as in the adult life (Dale, 1998). Wnts activate the ‘canonical signaling pathway’ by interacting with the 7TM receptors, Frizzled, and with low-density lipoprotein receptor-related proteins 5/6. Activation of the canonical Wnt pathway leads to inhibition of glycogen synthase kinase-3β (GSK-3β) by dissociating the enzyme from a multiprotein complex that involves axin, adenomatous polyposis coli, and β-catenin (Willert and Nusse, 1998) and via phosphorylation of GSK-3β on Ser-9 (Chen et al, 2000; Fukumoto et al, 2001). This results in the stabilization of the unphosphorylated form of β-catenin, which translocates into the nucleus, where it regulates the expression of Wnt-responsive genes that maintain neuronal homeostasis and support cell survival (Hinck et al, 1994; Aberle et al, 1997; Willert and Nusse, 1998). In vitro data have shown that neurons committed to die (e.g. neurons challenged with N-methyl-
Methods
Transient Focal Ischemia in Rats
We induced transient focal ischemia by infusing the potent vasoconstrictor, endothelin-1 (Et-1), in the brain parenchyma of male Wistar rats (270 to 310 g; Charles River, Calco, Italy). All rats were kept under environmentally controlled conditions (room temperature = 22°C, humidity = 40%) on a 12-h light/dark cycle with food and water ad libitum. Experiments were performed following the Guidelines for Animal Care and Use of the National Institutes of Health. Animals were anesthetized with a solution of ketamine hydrochloride (60 mg/kg) and xylazine (4 mg/kg) injected intramuscularly, and then placed on a Kopf stereotaxic apparatus. A 22-gauge stainless-steel cannula was implanted in the left hemisphere of animals 2.0 mm dorsal to the middle cerebral artery (MCA). The coordinates were as follows: 0.2 mm anterior, 5.2 lateral to bregma, and 6 mm below the skull, according to the atlas of Paxinos and Watson (1997; Sharkey et al, 1993; Moyanova et al, 2007). A 27-gauge dummy cannula was insert into the lumen to maintain pervious the guide.
After surgery, rats were removed from the stereotaxic apparatus, treated with an antibiotic, and then placed in individual cages with free access to water and standard pellets for 4 to 5 days. MCA occlusion was performed by microinjection of human/porcine Et-1 (Sigma, St Louis, MO, USA; 150 pmol in 3 µL of 0.9% saline). Injection was performed in the awake rats by means of a Hamilton 10 µL syringe connected to the injection needle by a short polyethylene tubing. Et-1 was given slowly for at least 3 mins and the needle was left in situ for a further 3 mins before being slowly withdrawn. Sham-operated rats were subjected to the same anesthesia and surgical procedure, but were injected with saline alone. Throughout surgery and during Et-1 or saline injection, body temperature was maintained at 37°C using a heating pad with a rectal probe. To select animals that had received the ischemic insult, we assessed motor deficits using the ‘hang reflex’ (PHR) test (Bederson et al, 1986; Moyanova et al, 2003). Animals were killed at 6 h (n = 5), 24 h (n = 5), 3 days (n = 8), and 7 days (n = 5) after injection, and brains were processed for histologic and immunohistochemical analysis. Different groups of animals received intraperitoneal (i.p.) injections of saline (n = 10) or lithium chloride (1 mEq/kg every 12 h; n = 10) (Sigma-Aldrich, Milan, Italy) for 7 days before the induction of focal ischemia and then for the following 3 days. Animals were killed 3 days after ischemia and the brains were processed for histologic analysis.
Permanent Focal Ischemia in Mice
Male C3H (Charles River, Calco, Italy) or mutant mice homozygous for a hypomorphic allele of Dkk-1 (doubleridge mice; provided by Miriam H Meisler), 10-week-old, weighing 22 to 24 g were used. Doubleridge mice are insertional mutant mice lacking of a transcriptional enhancer in the dkk-1 gene (Adamska et al, 2003; MacDonald et al, 2004). The animals were anesthetized with chloral hydrate (320 mg/kg, i.p.). With the aid of an operating stereomicroscope, an incision was made between the outer canthus of the eye and the external auditory meatus. The temporal muscle was bisected and retracted to expose the temporolateral surface of the skull. The MCA was exposed by means of a burr-hole craniotomy, performed with a dental drill (Welsh et al, 1987). A thin layer of bone was preserved to protect the dura mater and cortex surface from mechanical damage and thermal injury and remaining bone was removed. The MCA was occluded by electrocoagulation. The temporal muscle and then the skin incision were sutured (Backhauss et al, 1992). Sham-operated animals were subjected to the same anesthesia and surgical procedures, except the MCA occlusion. Animals were killed at 3 h (n = 5), 6 h (n = 5), 12 h (n = 5), 1 day (n = 5) or 3 days (n = 8), and were used for histologic/immunohistochemical analysis.
Different groups of animals (C3H and doubleridge mice) received i.p. injections of saline (n = 8) or lithium chloride (1 mEq/kg every 12 h; n = 8; Sigma-Aldrich) for 7 days before the MCA occlusion and then for the following 3 days. Animals were killed 3 days after ischemia and the brains were processed for histologic analysis.
Blue Evans Perfusion Study
Animals were perfused with the dye blue evans to compare the cerebrovascular anatomy of C3H mice (n = 5) and doubleridge mice (n = 5). Animals were anesthetized and perfused with 4% formalin followed by blue evans (1% in saline) via the left cardiac ventricle. After decapitation, the brains were carefully removed, and the vessels with their branches were examined under a microscope.
Histology
Brains were fixed in Carnoi, embedded in paraffin and sectioned at 10 µm. Sections were deparaffinized and processed for staining with thionin (Nissl staining for histologic assessment of neuronal degeneration). The analysis was performed on 10 µm sections regularly spaced every 550 µm through the extension of the ischemic region. The infarct volume was calculated by integrating the cross-sectional area of damage on each section and the distance between the various levels (Osborne et al, 1987). In each stained section, the necrotic area was identified and outlined at a magnification of ×2.5 and measured by the Scion Image software (NIH, Bethesda, MD, USA). The infarct volume (V) was calculated by using the following formula: V = Σ(Ai×T
Knowing that hypoxic brain tissue undergoes granulocyte infiltration (Noto et al, 2006), we stained deparaffinized sections with long Giemsa (Microstain Kit; Diapath, Martinengo, Italy) for the histologic assessment of blood cells. The extent of hypoxic volume was measured as described for the infarct volume.
Immunohistochemistry
Deparaffinized sections were soaked in 3% hydrogen peroxide to block endogenous peroxidase activity. Sections were treated with 10 mmol/L, pH 6.0, citrate buffer, and heated by microwave for 10 mins for antigen retrieval. The slides were allowed to cool for 20 mins in the same solution at room temperature and then washed in Tris-buffered saline. Rat brain sections were incubated overnight with rat monoclonal anti-Dkk-1 (1:10) (R&D System, Minneapolis, MN, USA), rabbit polyclonal anti-heat shock protein-70 (hsp-70; 1:100; Chemicon, Temecula, CA, USA) and rabbit polyclonal anti-β-catenin (1:20; Santa Cruz Biotechnology, Santa Cruz, CA, USA) antibodies, and then for 1 h with secondary biotinylated anti-rat or anti-rabbit antibodies (1:200; Vector Laboratories, Burlingame, CA, USA). 3,3-Diaminobenzidine tetrachloride was used for detection (ABC Elite kit; Vector Laboratories). Control staining was performed without the primary antibodies.
Mouse brain sections were incubated overnight with goat polyclonal anti-Dkk-1 (1:5; R&D System), rabbit polyclonal anti-β-catenin (1:5; Santa Cruz Biotechnology), and goat polyclonal anti-Bcl-2 (1:10; Santa Cruz Biotechnology) antibodies, and then for 1 h with secondary Cy3 anti-goat (1:300; Chemicon), fluorescein anti-rabbit antibodies (1:100; Vector Laboratories) or biotinylated anti-goat antibodies (1:200; Vector Laboratories). 3,3-Diaminobenzidine tetrachloride (ABC Elite kit; Vector Laboratories) was used for detection of Bcl-2. Control staining was performed without the primary antibodies.
Double fluorescence immunohistochemistry was performed incubating rat brain sections overnight with monoclonal rat anti-Dkk-1 (1:10; R&D System) and monoclonal mouse anti-glial fibrillary acidic protein (1:100; Sigma-Aldrich) or rabbit polyclonal anti-hsp-70 (1:100; Chemicon) antibodies, and then for 1 h with secondary Cy3 anti-rat (1:300 Chemicon) and fluorescein anti-mouse or anti rabbit (1:100; Vector Laboratories) antibodies.
Results
Temporal Profile of Ischemic Brain Damage in Rats Infused with Et-1 near to the MCA
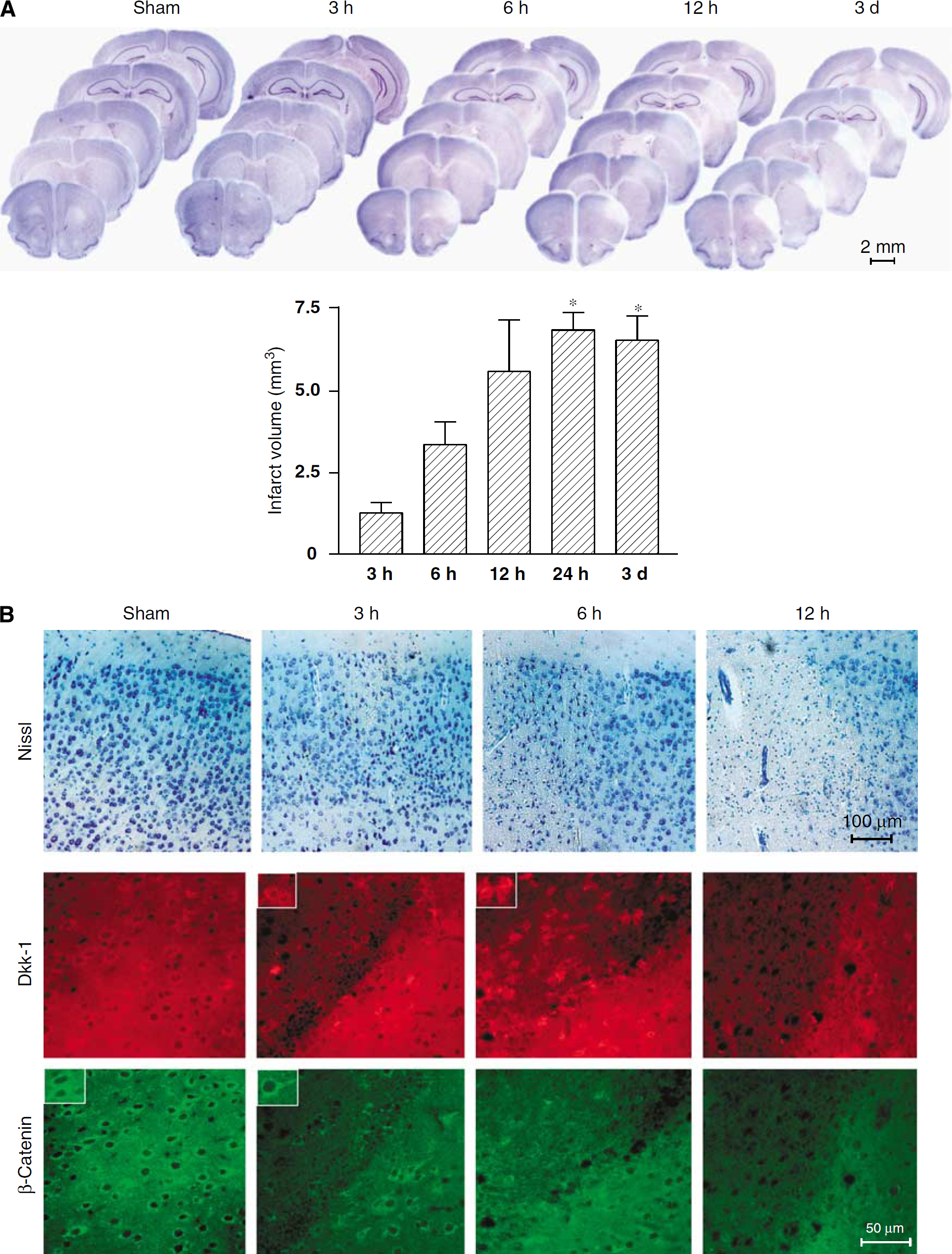
Unilateral infusion of Et-1 (150 pmol per 3 µL) near to the MCA caused a transient reduction of blood flow in the ipsilateral corticostriatal region. The reduction of the interstitial oxygen tension (pO2) was mirrored by a local infiltration of granulocytes at 6 to 24 h after Et-1 injection (Figure 1A). Nissl staining showed a remarkable increase in the infarct volume between 24 h and 3 days after Et-1 injection (Figure 1B). At 7 days, necrotic tissue was replaced by scared tissue (Figure 1B).
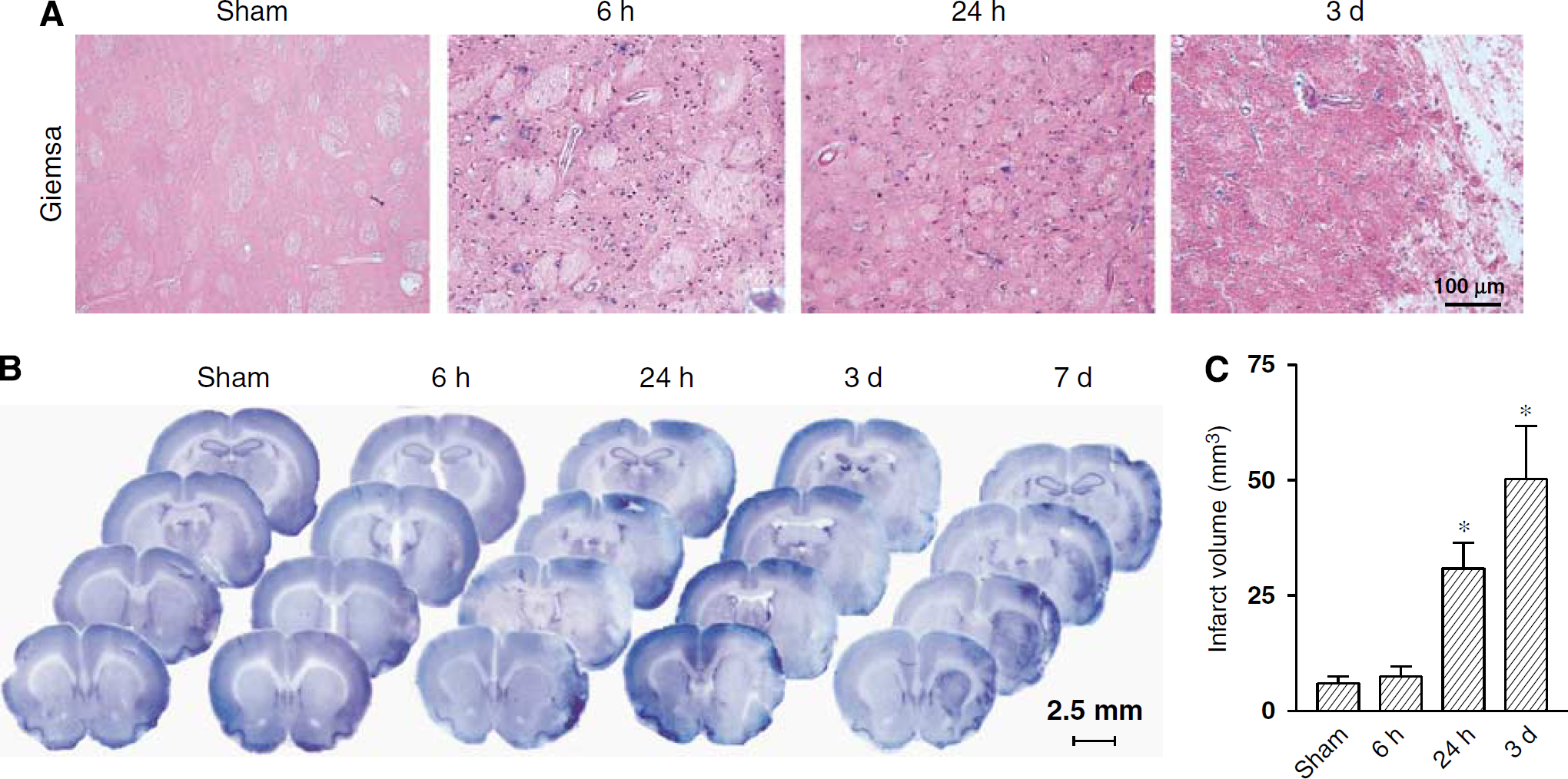
(
Induction of Dkk-1 and Reduced Expression of β-Catenin in the Ischemic Penumbra of Et-1 Injected Rats
Immunohistochemical analysis showed an early induction of Dkk-1 in the striatum 24 h after Et-1 injection, when the tissue showed massive infiltration of granulocytes, but no neuronal death (Figure 2A). At 3 days after Et-1 injection, Dkk-1-immunoreactive neurons were detected both in the ischemic core (not shown) and in the penumbra region, identified by the presence of cells expressing hsp-70 (Figure 2B). At higher magnification, Dkk-1 immunostaining was found outside the cell nucleus, which is consistent with the role of Dkk-1 as a secreted glycoprotein. Double fluorescent staining showed that Dkk-1 was absent in astrocytes (identified by glial fibrillary acidic protein immunostaining; Figure 2C) and was not induced in neurons expressing hsp-70 of the ischemic penumbra (Figure 2D). To examine whether induction of Dkk-1 was associated with inhibition of the canonical Wnt pathway, we assessed the expression of β-catenin at 3 days after Et-1 injection. We found a substantial reduction in neuronal expression of β-catenin in the same region of the ischemic penumbra in which we detected Dkk-1-immunoreactive neurons (Figure 3).
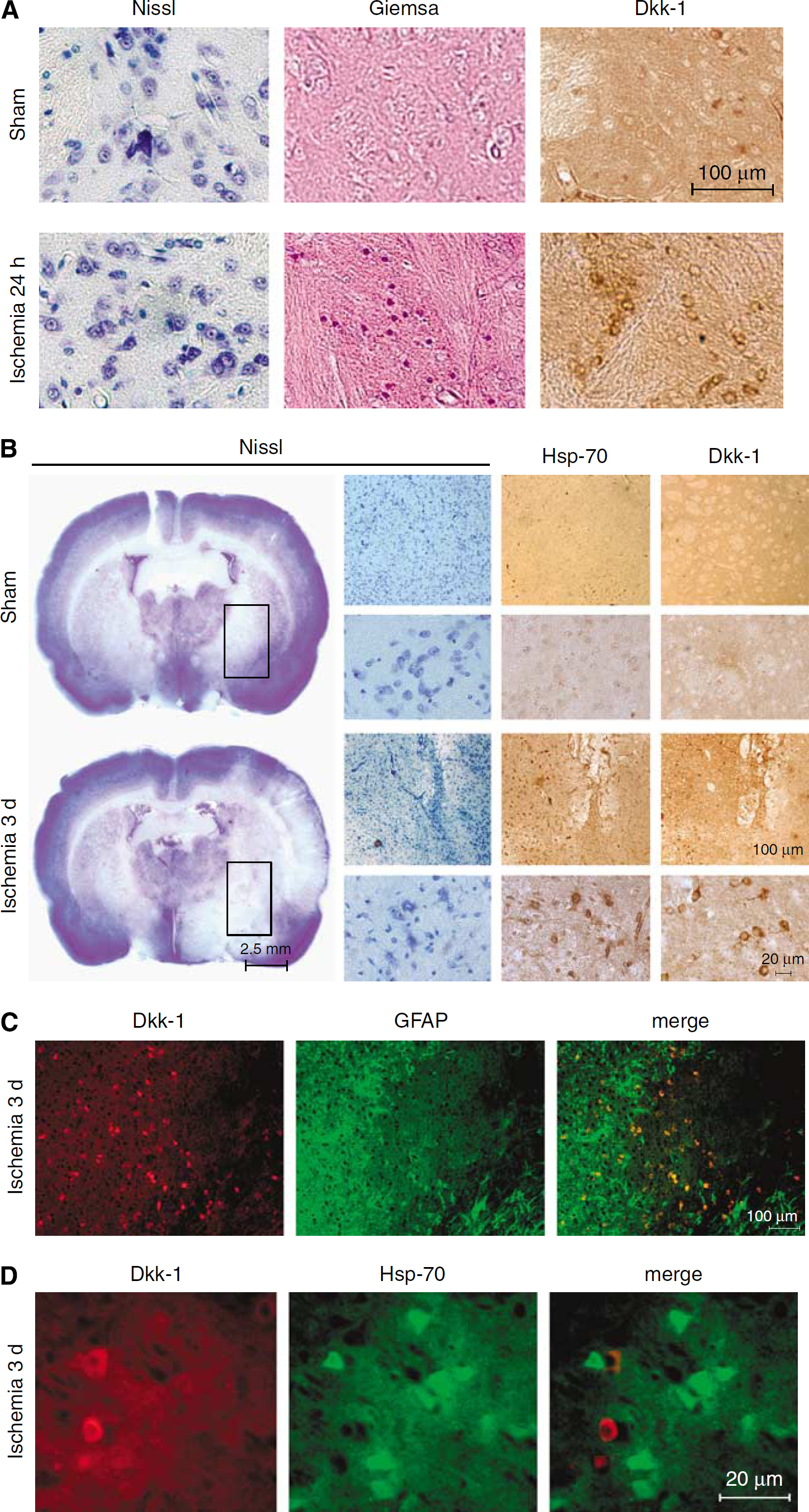
(
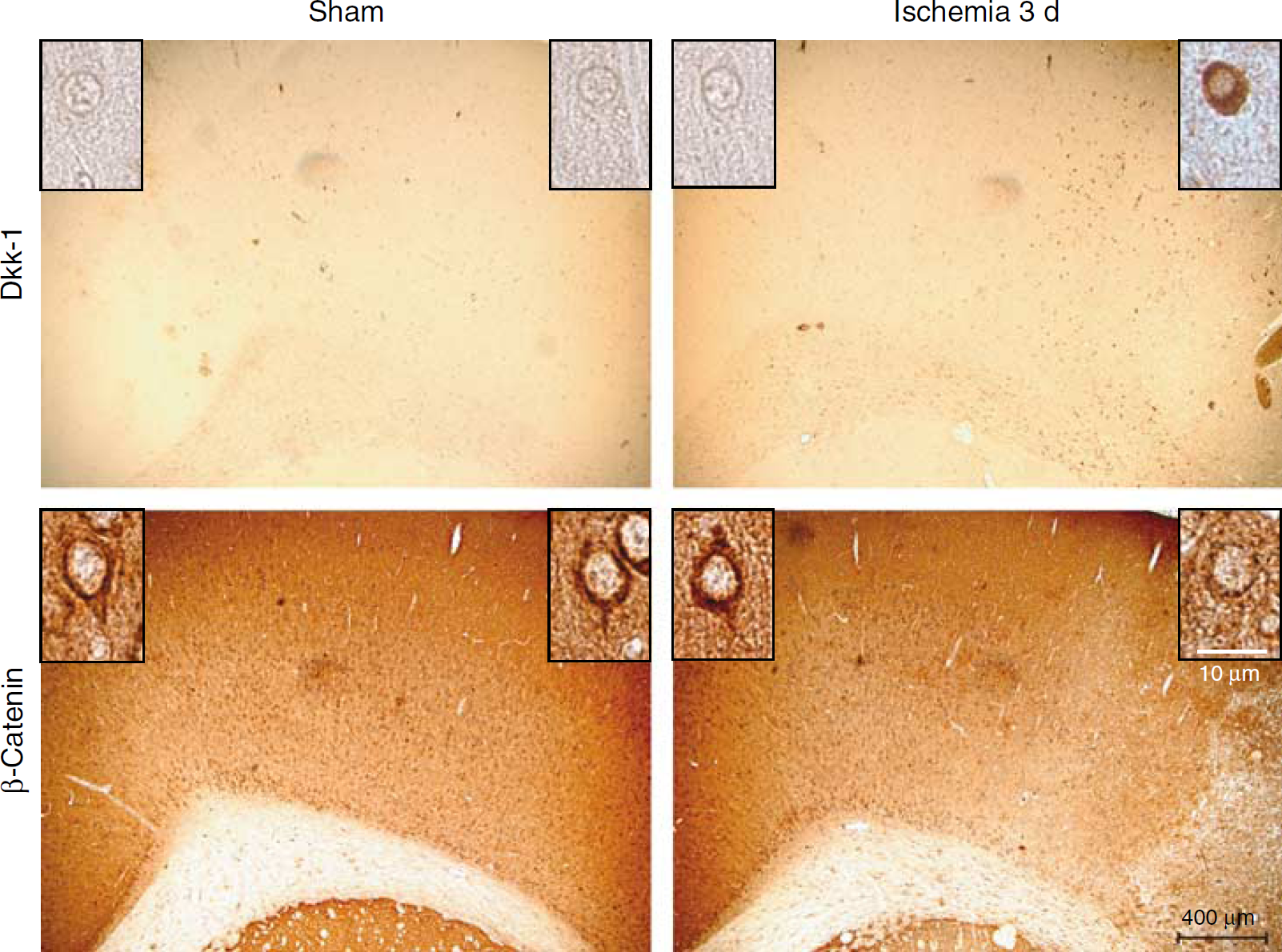
Dkk-1 and β-catenin expression in adjacent sections of the ischemic penumbra in 3 days (3 d) after Et-1 injection (150 pmol per 3 µL). Dkk-1 and β-catenin immunostaining is shown at higher magnification in the inserts.
Lithium Treatment Enhanced β-Catenin Expression and Protected Neurons against Ischemic Damage in Et-1 Injected Rats
We examined whether a pharmacological rescue of the canonical Wnt pathway could be protective against transient focal ischemia by treating rats with lithium ions (injected as lithium chloride every 12 h, at the dose of 1 mEq/kg, i.p., starting 7 days before Et-1 injection). Lithium ions are known to mimic the inhibitory action of Wnt on GSK-3β, thus rescuing the canonical Wnt pathway. When assessed at 3 days after Et-1 injection, lithium treatment prevented the reduction in β-catenin expression in neurons of the ischemic penumbra (Figure 4A), and was highly protective against ischemic neuronal damage (Figure 4B). Lithium treatment did not affect the induction of Dkk-1 in the ischemic penumbra (Figure 4A). This was expected because lithium rescues the canonical Wnt pathway acting downstream of the Dkk-1 blockade.
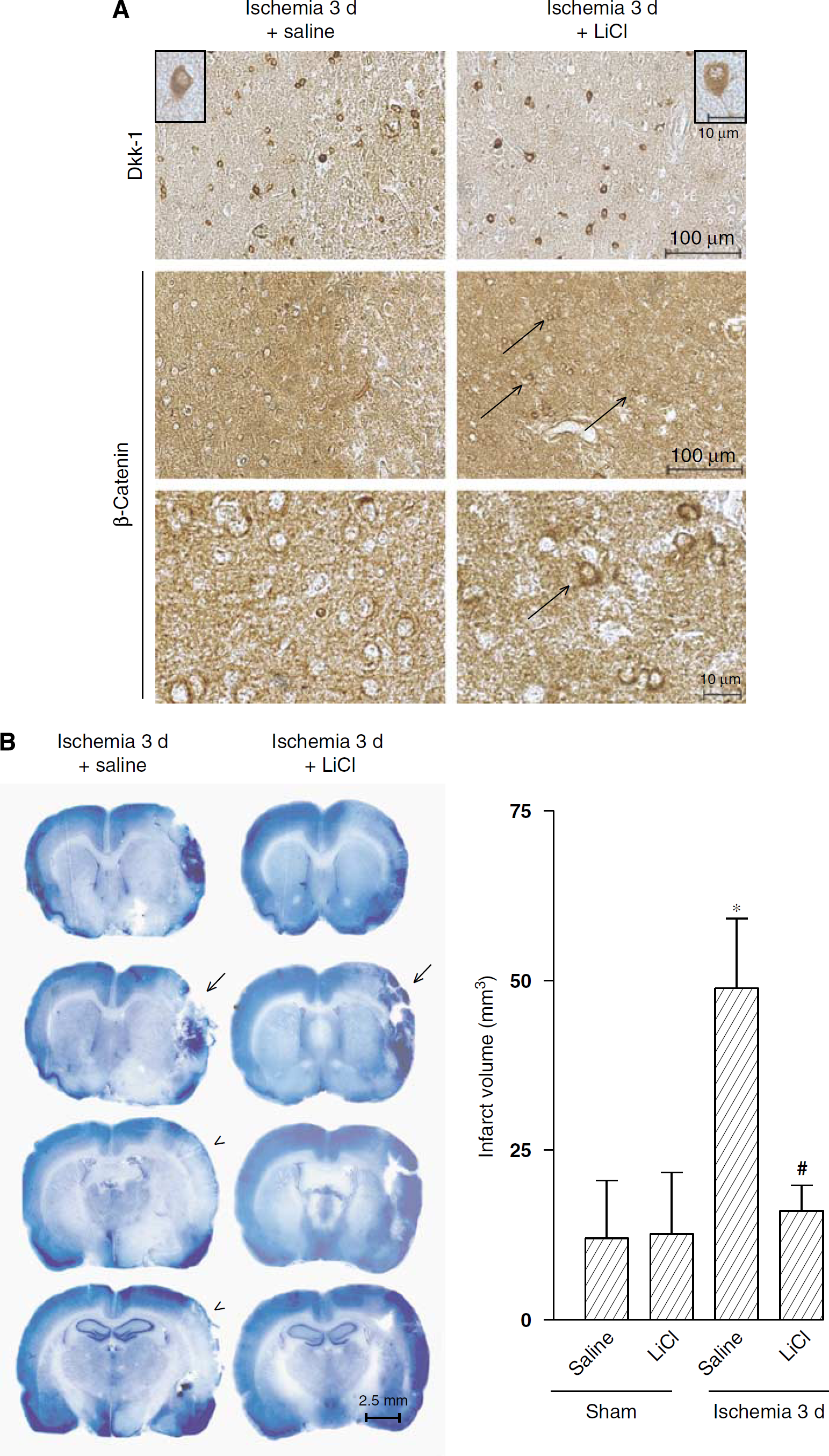
(
Induction of Dkk-1 Contributes to Neuronal Damage in a Mouse Model of Permanent Focal Ischemia
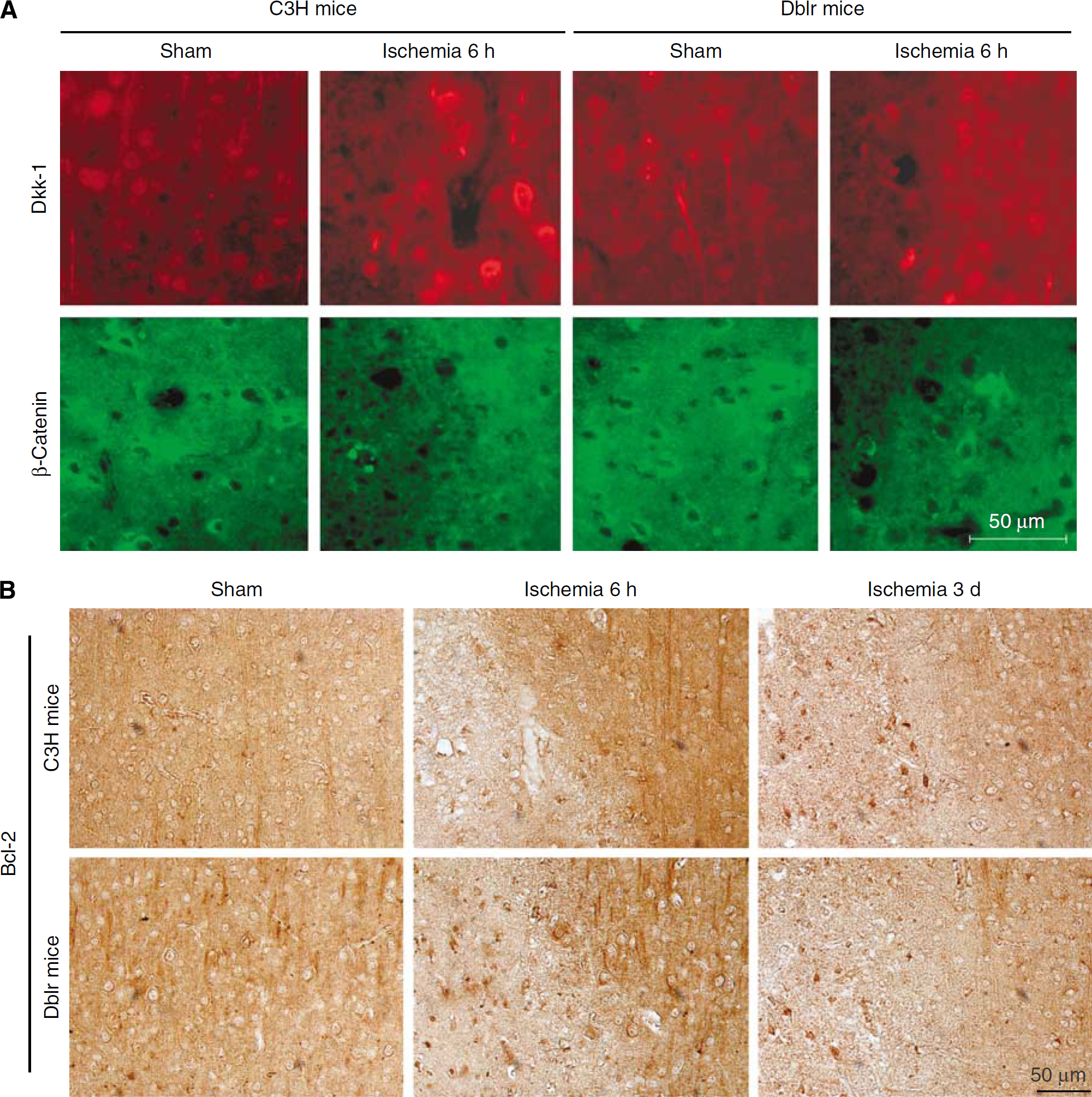
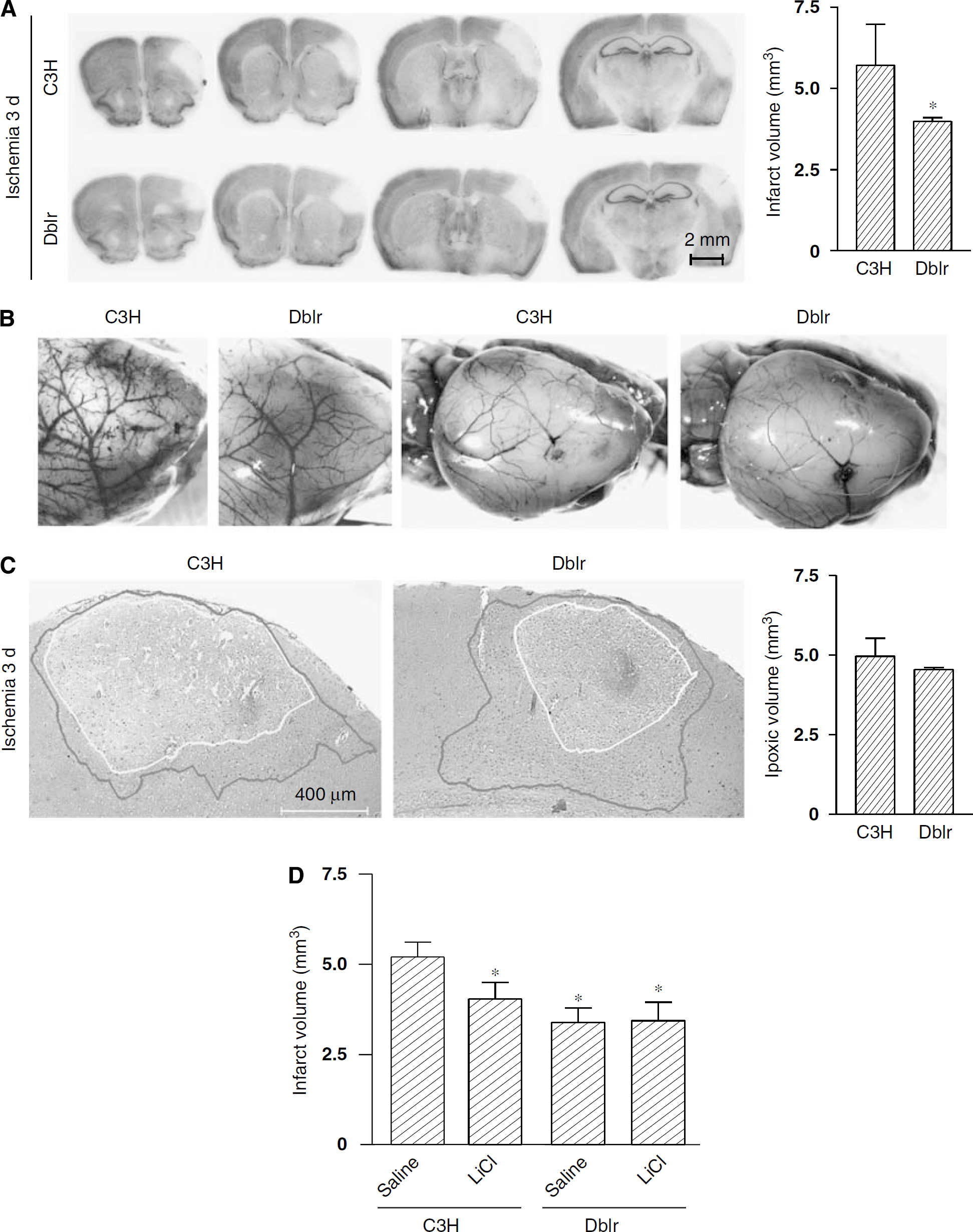
We extended the study of Dkk-1 to mice in which focal ischemia was induced by permanent MCA occlusion. We compared wild-type mice (C3H) with insertional mutant doubleridge mice that lack a transcriptional enhancer in the dkk-1 gene (Adamska et al, 2003; MacDonald et al, 2004). In C3H mice, MCA occlusion caused extensive ischemic damage of the cerebral cortex, which was already detectable at 3 to 6 h, and became complete at 24 h (Figure 5A). Immunohistochemical analysis showed a transient induction of Dkk-1 in neurons at early times (3 and 6 h) after MCA occlusion (Figure 5B). Dkk-1 induction was associated with a reduced expression of β-catenin in neurons in the same ischemic territory (Figure 5B). No induction of Dkk-1 was seen in response to MCA occlusion in doubleridge mice, as expected (Figure 6A). Doubleridge mice subjected to MCA occlusion showed a decrease reduction of β-catenin (Figure 6A) associated with a greater induction of the antiapoptotic factor, Bcl-2 (Figure 6B), and a reduced infarct volume as compared with ischemic C3H mice (Figure 7A). Neuroprotection did not result from a reduced hypoxic volume and did not depend on anatomic abnormalities of the MCA in doubleridge mice (Figures 7B and 7C). Treatment with lithium ions (injected as lithium chloride every 12 h, at the dose of 1 mEq/kg, i.p., starting 7 days before MCA occlusion) was partially protective against permanent focal ischemia in wild-type mice (Figure 7D). The action of lithium was occluded in doubleridge mice, where lithium treatment did not further reduce the infarct volume after MCA occlusion (Figure 7D).
(
(
(
Discussion
Our data demonstrate that a rapid induction of the secreted glycoprotein, Dkk-1, with the ensuing inhibition of the canonical Wnt pathway, is associated with and causally related to neuronal damage in rat and mouse models of focal ischemia. Dkk-1 has a prominent role during development, and is expressed at very low levels in the adult brain (Glinka et al, 1998; Diep et al, 2004). Thus, the induction of Dkk-1 in ischemic neurons provides a remarkable example of how a reexpression of a developmental protein can contribute to processes of neuronal death. We used a rat model of transient focal ischemia based on the intracerebral infusion of the potent vasoconstrictor, Et-1, near to the MCA. This model (see Moyanova et al, 2007 for a detailed characterization) recapitulates features of thromboembolic stroke in humans, and is technically advantageous because it is associated with a low rate of mortality and allows a reproducible assessment of pathologic processes associated with focal ischemia. Remarkably, Dkk-1 induction was not restricted to the ischemic core but was also observed in neurons of the ischemic penumbra that can be efficiently targeted by neuroprotective strategies. For technical reasons, we could not assess the expression of Dkk-1 and β-catenin in the same cell. However, a reduction of neuronal β-catenin levels was consistently found in the same region of the ischemia penumbra in which we observed a robust induction of Dkk-1. Thus, it is likely that Dkk-1 induction was associated with the inhibition of the canonical Wnt pathway, leading to an enhanced degradation of the downstream protein, β-catenin (Aberle et al, 1997; Hart et al, 1998). This could have limited the amount of β-catenin available for nuclear translocation (Levina et al, 2004), thus depriving neurons from the trophic support provided by the transcriptional program triggered by β-catenin. A causal role for Dkk-1 in ischemic neuronal damage was supported by the protective action of lithium in the Et-1 model of focal ischemia. Lithium treatment did not affect Dkk-1 induction, but prevented the loss of β-catenin in ischemic neurons. This was predictable because lithium is known to inhibit GSK-3β, thus rescuing the canonical Wnt pathway downstream of the Dkk-1 blockade (Klein and Melton, 1996). It should be highlighted, however, that lithium exerts pleiotropic effects in neurons, and that neuroprotection may result from the convergence of multiple intracellular processes (Lenox and Wang, 2003). Our data are in line with the protective action of lithium shown in models of focal and global ischemia (Nonaka and Chuang, 1998; Ren et al, 2003; Cappuccio et al, 2005; Yan et al, 2007). An interesting finding was that expression of Dkk-1 and hsp-70 was mutually exclusive in neurons of the ischemic penumbra. Hsp-70 is protective against hypoxic/ischemic neuronal death (Uney et al, 1994; Amin et al, 1996; Papadopoulos et al, 1996; Fink et al, 1997; Xu and Giffard, 1997; Rajdev et al, 2000; Lee et al, 2001) and enhances the degradation of the tumor-suppressing protein, p53 (Zylicz et al, 2001), which has been implicated in the pathophysiology of ischemic neuronal damage (Watanabe et al, 1999; Leker et al, 2004). Interestingly, the dkk-1 gene is a transcriptional target for p53, and increases in p53 levels drive the expression of Dkk-1 in cultured neurons challenged with βamyloid (Caricasole et al, 2003). A possible scenario is that DNA damage increases the expression of p53 in neurons of the ischemic penumbra, and p53 in turn drives the induction of Dkk-1 and the resulting inhibition of the canonical Wnt pathway. This pathologic cascade may be hampered in hsp-70-expressing neurons because of the inhibitory effect of hsp-70 on p53 expression. An intriguing finding is that lithium treatment upregulates hsp-70 in ischemic neurons as a result of GSK-3β inhibition (Ren et al, 2003; Xu et al, 2006; Bian et al, 2007). This suggests a dual mechanism whereby lithium protects ischemic neurons by interfering with the canonical Wnt pathway. From one side, lithium would protect Dkk-1-expressing neurons by rescuing the canonical Wnt pathway; from the other side, lithium treatment would prevent Dkk-1 induction by enhancing the expression of the protective protein, hsp-70. The use of mice overexpressing or lacking hsp-70 is required to examine these mechanisms in detail.
We extended the study to a mouse model of permanent focal ischemia, which allowed us to directly examine whether expression of Dkk-1 was causally related to ischemic neuronal death. We could address this issue by comparing wild-type mice with doubleridge mice, which were identified in a screen for recessive, transgene-induced insertional mutants (Adamska et al, 2003). In the doubleridge mutation, insertion of a 50-kb transgene composed of tandem copies of a 6.5-kb construct is accompanied by deletion of 60-kb of genomic DNA from the insertion site. The insertion site is located 150-kb downstream of the dkk-1 gene resulting in the loss of a transcriptional enhancer in the dkk-1 gene (Adamska et al, 2003; MacDonald et al, 2004). Thus, doubleridge mice show an intact intron/exon structure of dkk-1, but a reduced gene expression during development (MacDonald et al, 2004) or in response to environmental cues (Busceti et al, 2008). As opposed to wild-type mice, doubleridge mice did not respond to permanent focal ischemia with an induction of Dkk-1 and a reduction in β-catenin levels, and showed a 45% reduction in the infarct volume as compared with wild-type mice. The decrease reduction of β-catenin in doubleridge mice was associated with a greater induction of the antiapoptotic protein, Bcl-2, which is known to be regulated by β-catenin (McEntee et al, 1999; Li et al, 2007). That a rescue of the canonical Wnt pathway accounted for the relative resistant of doubleridge mice to permanent focal ischemia was supported by evidence that the protective effect of lithium treatment was no longer visible in these mice.
Taken together, our data increase the attractive possibility that Dkk-1 antagonists or drugs that rescue the canonical Wnt pathway (e.g. GSK-3β inhibitors with a better profile of specificity and tolerability than lithium ions) may protect neurons against focal ischemia and limit the extension of damage across the ischemic penumbra. The development of Dkk-1 antagonists is under progress and is allowed by the evidence Dkk-1 binds to a region of low-density lipoprotein receptor-related proteins 5/6 different from the region that containing the binding site for Wnt glycoproteins (Semënov et al, 2001). Dkk-1 antagonists can be viewed as promising neuroprotective agents because they specifically address a pathologic mechanism and, as such, should not interfere with physiologic synaptic transmission (as opposed to glutamate receptor antagonists) or with intracellular mechanisms that control neuronal homeostasis.