
Abstract
The flow of new information on gene expression related to apoptosis has been relentless in the last several years. This has also been the case with respect to gene expression after cerebral ischemia. Many of genes associated with an apoptotic mode of cell death have now been studied in the context of experimental cerebral ischemia from the immediate early genes through modulating genes such as bcl-2 to genes in the final execution phase such as interleukin-1β converting enzyme (ICE)-related proteases. It was impossible to adequately cite all primary reports on these subjects. However, many excellent reviews have appeared in the last year, which together, cover all these areas of interest. In this review, we have elected to cite only reports published since January 1996 and use an extensive collection of reviews (indicated in italics) to guide the reader to the earlier literature. Our intent is to provide the reader with a timely and useful analysis of the current state of the art. It is hoped that this approach does not cause offense with our colleagues whose contributions before 1996 laid the foundation for much of this work.
THE YEAR IN REVIEW: 1996
A general review very useful to those just starting to seek an understanding of apoptosis, which also includes details of protease cascades and therapeutic possibilities, was written by Duke et al. (1996). In summarizing signal transduction involved with apoptosis, McConkey and Orrenius (1996) highlighted the difficulty with generalizations, and emphasized the importance of the context in which signals are processed by cells by the statement “signals that promote apoptosis in one model can suppress cell death in another.” Cells are overproduced during development to form tissues and then culled by programmed cell death to adjust numbers and sculpt the final form, a process eloquently derived by Jacobson et al. (1997).
The molecular regulation of apoptosis was reviewed from different perspectives by Desbarats et al. (1996), Hale et al. (1996), Nagata (1997) and Vaux and Strasser (1996) with emphasis on the conserved phylogenetic nature of the cell death machinery, and its interlocking with the control of cell proliferation in conjunction with influence of extracellular factors such as cytokines. The degradative enzymes such as the ICE-like proteases and endonucleases were specifically covered by Montague and Cidlowski (1996), Mundle et al. (1996), Nicholson (1996) Schwartz and Milligan (1996), and Ashkenhas and Werb (1996). A nomenclature for the human homologs of the ICE/Ced-3 proteases, now called caspases (cysteine proteases cleaving after an asp residue) has been defined based on phylogenetic relationships (Alnemri et al., 1996). The Bcl-2 family of proteins, containing apoptosis, inhibitors and inducers, was analyzed by Yang and Korsymeyer (1996) and Kroemer (1997) while Bredesen (1996a,b) specifically addressed the Bcl-2 family in neurons.
Several lines of evidence point to a central role for mitochondria in effecting apoptotic cell death. The disruption of the mitochondrial transmembrane potential and opening of the permeability transition pore early in apoptosis are linked with release of some mitochondrial substance that appears capable of inducing apoptotic characteristics in cell free systems (Kroemer et al., 1997). A clinical assessment of the applicability of this basic knowledge, particularly of the signalling of cell death from the TNF family of receptors, left the reader with the understanding of the work still to be done before practical benefit will be possible (Rudin and Thompson 1997), and concluded with “The era of widespread clinical implementation of apoptotic modulation in the treatment of disease has not yet arrived.”
How this general understanding of apoptotic cell death applies to neurons in particular was covered by Bredesen (1996a,b), Linnik (1996), Mattson and Furukawa (1996), Ross (1996), and Waters (1996). These texts cover the definitions of apoptosis, the elegant genetic work performed on cell death in the nematode C. elegans, genes that promote cell death (eg, jun, myc, p53), genes that oppose cell death (eg bcl-2, IAP), neuronal death during development, and an evaluation of potential pharmacological and therapeutic intervention strategies. The specific involvement of apoptosis after cerebral ischemia was reviewed in several publications (Charriaut-Merlangue, et al., 1996a; Choi, 1996; Chopp and Li, 1996; Edwards and Mehmet, 1996; Ferrer 1996; Mehmet and Edwards, 1996) that cover some of the same ground as the reviews above but also delve into the thorny issue of neuronal necrosis. The central role for excitotoxicity-inducing cellular mortality via changes in calcium transients was also considered (Bennett and Huzlin, 1996; Choi 1996) and links were made with changes in the mitochondrial permeability transition pore (Kristian and Siesjo 1996; Siesjo and Siesjo 1996). Immediate early gene expression such as fos and jun in brain (Akins et al., 1996; Herzog and Morgan, 1996) and other stress genes such as heat shock proteins (Massa et al., 1996) have also become a major area of investigation.
Another rapidly evolving area concerning gene expression induced by cerebral ischemia relates to neuroinflammation, cytokines and adhesion molecules (Arvin et al., 1996; Kim, 1996; Matuso et al., 1996), and includes interleukin-1 (Betz et al., 1996). Finally, there is no doubt as to the importance of oxidative damage caused by hydroxyl, superoxide, or nitric oxide radicals in the evolution of the ischemic infarct and general neurotoxicity (Chan, 1996; Dawson and Dawson 1996; Hosler and Brown 1996; Iadecola, 1997; Szabo 1996), and it is now likely that future therapeutic treatments for stroke will have an antioxidant component. It is here that we come full circle because of the importance of oxidative mechanisms in controlling apoptosis (Bredesen 1996b; Hale et al., 1996; McConkey and Orrenius, 1996; Mattson and Furukawa 1996). An overall scheme defining many of the intracellular factors that influence the survival of a cell after a stroke are indicated in Fig. 1.
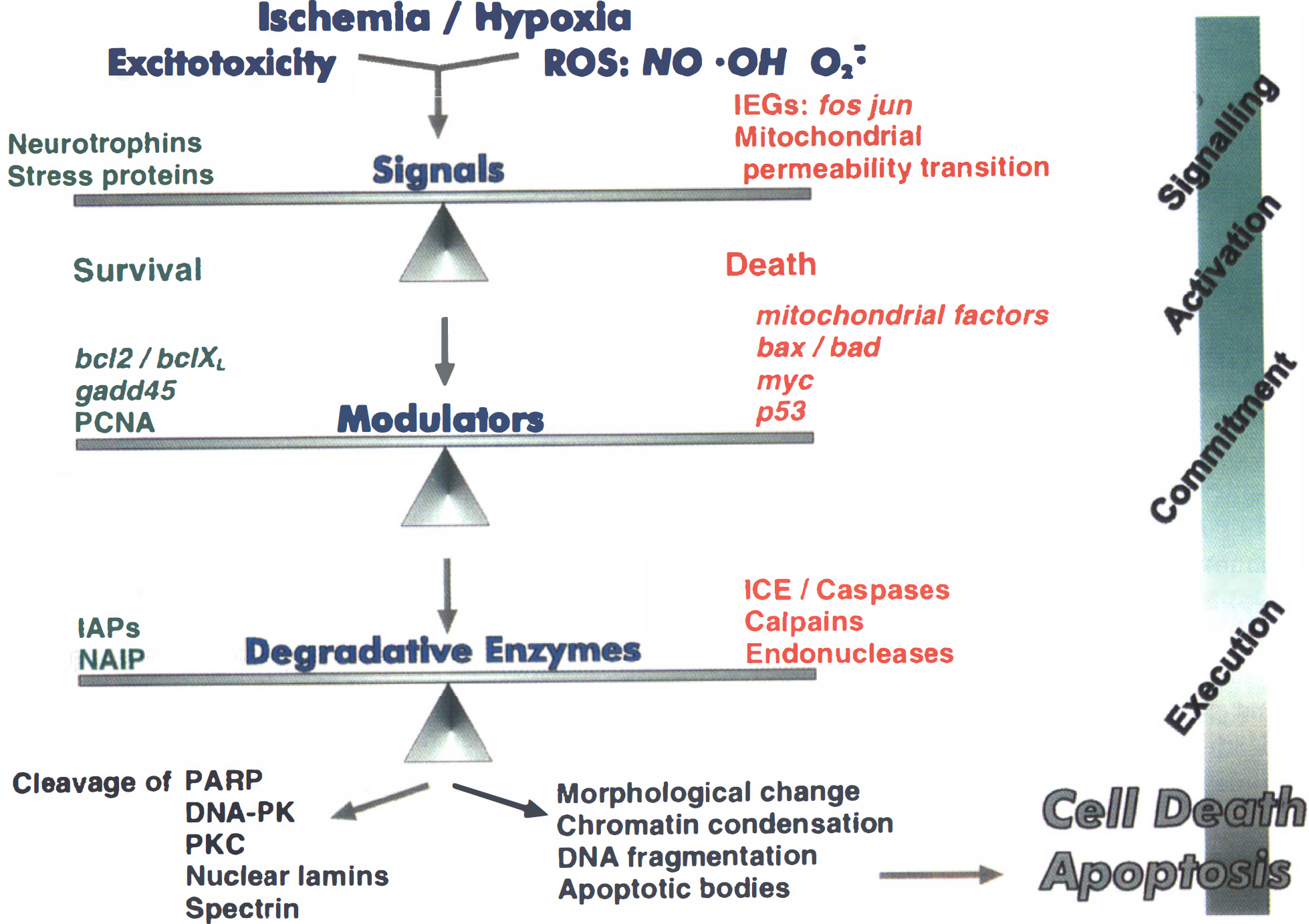
There are several steps between the initial ischemic/hypoxic insult and frank neuronal death. Within this cell death cascade, there are several molecules and proteins that facilitate neuronal survival and compete with factors that contribute to the cell death cascade. Ultimately, the balance between survival factors and death factors determines the fate of the cell. Pharmacological approaches to stroke can be directed at facilitating the survival factors or inhibiting the cell death machinery, with the goal of preserving neurons until reperfusion and reoxygenation of the tissue can be established.
THE Bcl-2 GENE FAMILY: APOPTOSIS INHIBITORS AND INDUCERS
The Bcl-2 family of proteins are homologous to the ced-9 gene product in C. elegans that has the remarkable property of inhibiting developmental cell death, particularly in the nervous system. There are now several members of the Bcl-2 family (Table 1) that are structurally, but not functionally, homologous. In an intriguing turn of evolution, the original ced-9 gene has evolved into a family of genes with specific members that inhibit apoptosis and others that promote apoptosis.
Bcl-2 protein family
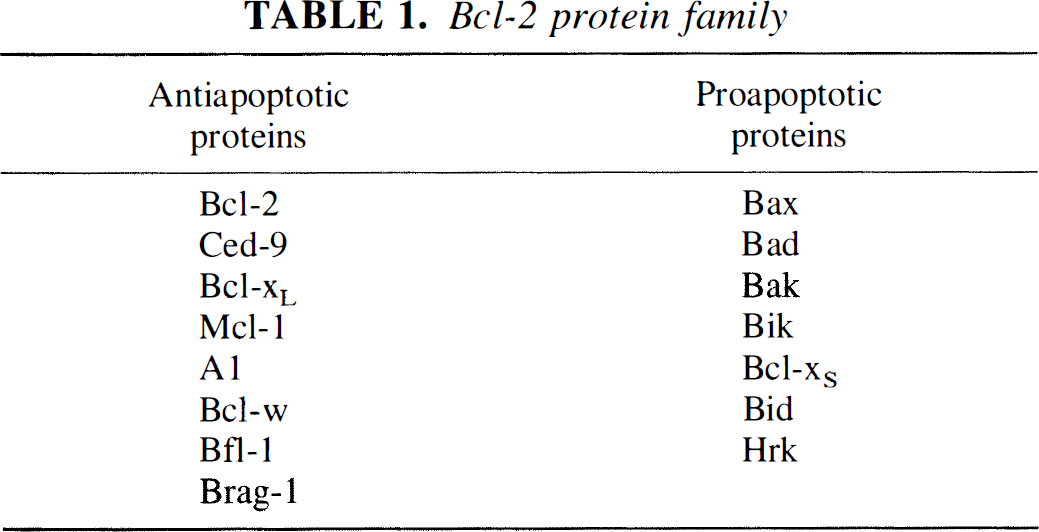
The analysis of the bcl-2 gene family in ischemic stroke is still in its infancy, but the initial results suggest an important role in determining neuronal survival. In vitro studies showed that Bcl-2 protected cells against a wide variety of insults, including excitotoxicity, elevated calcium, lipid peroxidation, and free-radical damage. The important role of these factors in ischemia suggested that increased Bcl-2 expression might be protective in animal models of stroke. A neuroprotective effect of Bcl-2 has been shown in transgenic animals that overexpress Bcl-2, or when viral vectors are used to overexpress Bcl-2 in focal, or global ischemia (Choi, 1996; Chopp and Li, 1996; Linnik 1996; Lawrence et al., 1996).
Several studies have analyzed the expression of the proapoptotic and antiapoptotic members of the bcl-2 gene family after focal and global cerebral ischemia. To date, these studies have specifically concentrated on the protective proteins Bcl-2 and Bcl-xL and the death enhancing Bax protein as these proteins have been shown to be important determinants of neuronal apoptosis in the developing brain (Deckwerth et al., 1996; Shindler et al., 1997). In general, the results from stroke studies have been consistent with the known functions of these proteins. Specifically, the proapoptotic Bax protein is upregulated in neurons that are destined to die in both global ischemia (Chen et al., 1996; Hara et al., 1996) and focal ischemia (Gillardon et al., 1996b) while its expression remains constant in neurons that survive the ischemic insult. The expression of Bcl-2 and Bcl-xL after ischemia is also consistent with their known function as inhibitors of apoptosis. Both Bcl-2 and Bcl-xL are persistently expressed in neurons that survive in focal ischemia (Gillardon et al., 1996b; Asahi et al., 1997) and global ischemia (Honkaniemi et al., 1996a; Chen et al., 1996 and 1997). The differential expression of these family members is most remarkable in the global ischemia models where the cell death is localized to the vulnerable CA1 hippocampal neurons. Thus, Bax overexpression is solely localized to the CA1 neurons, while sustained Bcl-2 and Bcl-xL expression is observed in CA3 and the dentate gyrus, but not in the CA1 neurons.
The specific mechanism(s) by which Bcl-2-related proteins influence survival in cerebral ischemia remain to be determined (see Box 1). While most candidate mechanisms have not been analyzed in stroke, the significance of dimerization within the Bcl-2 family is suggested in studies describing the expression of Bcl-2, Bcl-xL, and Bax after ischemia. For example, the persistence of Bax expression with reduction of Bcl-2 and Bcl-xL in the CA1 region after global ischemia would enhance the formation of proapoptotic Bax homodimers. However, direct quantitation of the hetero- and homodimerization profile in normal and ischemic hippocampal neurons remains to be established.
Studies on the transcriptional regulation of the bcl-2 genes has led to interesting descriptions of known apoptotic factors influencing transcription in this gene family. For example, the p53 tumor suppressor protein can increase the expression of bax mRNA, possibly as an adjunct mechanism for inducing cell deletion. The potential relevance of this mechanism to excitotoxicity was shown by Hughes et al. (1996) who showed that a quinolinic excitotic lesion in rats induced p53 expression that led to a increase in bax mRNA.
The search for the mechanism(s) underlying the antiapoptotic effects of Bcl-2 have been underway since the discovery of the Ced-9 gene in C. elegans. One early observation was the demonstration of single transmembrane region driving the subcellular localization of Bcl-2 primarily to mitochondrial and nuclear membranes. This suggested that the protective effect of Bcl-2 was related to the preservation of mitochondrial function and/or nuclear integrity. However, molecular studies deleting the transmembrane region proved that the membrane localization was not absolutely necessary for the antiapoptotic activity. In addition, Bcl-2 was able to block apoptosis in cells that did not contain mitochondria, again suggesting that mitochondrial membrane localization was not critical for function (Yang and Korsmeyer, 1996; Zhu et al., 1996). The search for a mechanism related to Bcl-2 was further frustrated by the inability to demonstrate any effect of Bcl-2 on normal organelle function, including mitochondrial respiration or nuclear transport. The data suggested that Bcl-2 was an innocuous protein with no readily discernible function except the potent and promiscuous prevention of apoptotic and even necrotic cell death in many cell types and cell death models (Bredesen, 1996a).
Perhaps the largest single gap in the literature concerning Ced-9 and Bcl-2 has been the lack of a firm connection between Bcl-2 and the other critical component of the apoptotic cascade, the Ced-3/caspase family of proteases. In C. elegans, the caspase Ced-3 is absolutely required for programmed cell death. However, until recently there was no direct biochemical connection between the proteolytic function of Ced-3/ICE and Ced-9/Bcl-2. Several researchers are now beginning to elucidate the potential mechanism(s) underlying the antiapoptotic effect of the Bcl-2 family of proteins. In addition, there is new data defining the relationship between the caspases and the bcl-2 proteins (see Box 1). Much of this data suggests an important role for protein-protein interactions and mitochondria localization of Bcl-2. However, this data is very recent and the interrelationship between the mechanisms is not always obvious. In addition, the specific mechanisms that are most relevant to ischemic neuronal death remain to be determined.
How does Bcl-2 inhibit apoptosis?

Bcl-2 inhibits both apoptosis and necrosis. However, Bcl-2 expression has little effect on metabolic function in normal cells. Several lines of investigation are now suggesting mechanisms by which Bcl-2 might prevent cell death and many of these mechanisms may be applicable to ischemic cell death.
At present, the majority of dimerization data can best be fit to two potential model (Gajewski and Thompson, 1996). In the first model, the formation of toxic Bax homodimers is dominant, and Bcl-2/Bcl-xL negatively regulate cell death by inhibiting the formation of toxic Bax homodimers. Alternatively, Bcl-2/Bcl-xL dimers may be dominant repressors of cell death, and heterodimerization with Bax or Bad inhibits the ability of these molecules to prevent apoptosis. In addition to preventing permeability transition, Bcl-2 also enhances the calcium uptake capacity of the mitochondria and provides mitochondria with a greater resistance to Ca++ induced respiratory injury (Murphy et al., 1996). All of these factors suggest that Bcl-2 stabilizes mitochondria in apoptosis and increases the capacity of a cell to sequester the increased calcium seen in ischemic injury. There are also observations suggesting that Bcl-2 prevent the release of an apoptogenic protease from the mitochondria, perhaps as a result of the permeability transition (Martins and Earnshaw, 1997; Susin et al., 1996). However, the structure of this protein has yet to be identified.
CASPASES: THE Ced-3/ICE FAMILY OF EXECUTIONERS
The Ced-3/ICE family of cysteine proteinases, now called caspases (Alnemri et al., 1996) have received a great deal of attention because of their key role as a final common executioner of apoptosis (Schwartz and Milligan, 1996; Nicholson, 1996) (Table 2). The caspases are expressed as proenzymes that undergo proteolytic processing and subunit rearrangement to yield active enzyme. Specifically, the proenzyme is cleaved at Asp-X junctions to yield a large subunit (p20) and a small subunit (p10). The crystal structure of caspase-1 suggests that the large and small subunits heterodimerize and two independent p10/p20 heterodimers associate to form a (p10/p20)2 tetramer with two active sites at opposite ends of the complex (Schwartz and Milligan, 1996; Nicholson, 1996). Thus, the caspases are well situated to regulate apoptotis since they are normally expressed in an inactive configuration and activation only occurs when an apoptotic signal is received.
The human caspase family*

ICE, interleukin- 1β converting enzyme.
Alnemri et al. (1996) classified the human homologs of the ICE/Ced-3 family of proteases as capases. Specific caspase members are identified along with their previous designations.
The caspase family contains several members with unique tissue distribution, substrate specificity, and activation criteria. Particular attention has been directed to the caspase-3 (CPP32) homolog because of its profile of preferred substrates that are clearly different than that of caspase-1 (ICE) (Schwartz and Milligan, 1996; Nicholson, 1996). For example, caspase-1 is very potent at converting pro-IL-1β to IL-1β, whereas caspase-3 has relatively little affinity for this substrate. In contrast, caspase-3 has a much higher affinity relative to caspase-1 for the DNA repair enzyme, poly (ADP-ribose) polymerase (see Box 2). In addition, caspase-3 has the greatest degree of homology to the Ced-3 cell death gene in C. elegans.
Compelling data linking caspase-3 to neuronal apoptosis comes from caspase-3 genetic knockout animals (Kuida et al., 1996). Although deleting the caspase-3 gene resulted in 100% lethality by week 3 of fetal development, these animals showed significant neuronal hyperplasia. These supernumerary cells were postmitotic, terminally differentiated, and were defined as neurons by histological and immunohistochemical methods. Interestingly, the caspase-3 knockout phenotype was largely confined to the nervous system as no abnormalities were identified in several other organ systems.
Caspase activation in ischemia
Currently, the most direct method for implicating the caspases in cell death is to use selective caspase inhibitors in a cell death model. Reasonable degrees of selectivity can be obtained with tetrapeptide inhibitors, while shorter peptides display a more promiscuous enzyme inhibition profile. The commercially available inhibitors typically use a carboxyl terminal aldehyde or ketone functionality as the activated warhead. To generate specificity, the active functionality is coupled to an amino acid recognition sequence that is based on known substrate cleavage sites for the specific caspase (Nicholson, 1996). Thus, the YVAD aldehyde shows > 10,000-fold specificity for caspase-1 (Ki ⩽ 1 nmol/L) over caspase-3 (Ki = 12 µmol/L). In contrast, the DEVD aldehyde is a subnanomolar inhibitor of caspase-3, although it also shows reasonable activity against caspase-1. The amino terminus in these inhibitors is critical for distinguishing between caspase family members and truncation to a tripeptide results in the loss of the ability to distinguish between family members.
Nuclear integrity and the caspases

Caspase activation is integral to most, if not all forms of apoptosis. Recent data has suggested a link between caspase activation and the changes in nuclear morphology that are a hallmark of apoptosis. Several important nuclear proteins have been identified as caspase substrates and the degradation of these proteins may contribute to chromatin condensation and/or DNA fragmentation. Some of the caspase substrates that are localized to the nucleus are identified below.
Two recent studies have reported that intracerebroventricular administration of caspase inhibitors can protect the brain from ischemia. Loddick et al. (1996a) showed that intracerebroventricular administration of the irreversible caspase inhibitor z-VAD-DCB was protective in a focal cerebral ischemia model. Hara et al. (1997) used tetrapeptide inhibitors to show neuroprotection with a caspase 1 specific inhibitor (YVAD-chloromethylketone) and a caspase-3 inhibitor (DEVD-fluoromethylketone). The pharmacology of this protection is not completely resolved because the caspase-3 inhibitor (which also inhibits caspase-1 to a lesser extent) seemed to be less efficacious than the caspase-1 specific inhibitor used in this study (Hara et al., 1997). In the case of the caspase-1 inhibitor, a component of the protection may be caused by inhibition of 1L-1β production, because the caspase-1 inhibitor, but not the caspase-3 inhibitor, attentuated IL-1β levels in the brain at 30 minutes after ischemia (see below).
Caspase-1/IL-1β in ischemia
The data linking caspase activation to ischemia-induced apoptosis is primarily based on the pharmacological profile of caspase inhibitors in the ischemia models. However, this is an extremely active area of research and several studies can be expected to appear in the coming months linking caspase activation to ischemic neuronal death. Meanwhile, there is substantial data linking the preferred substrate of caspase-1, IL-1β, to cell death in ischemia. Therefore, it is important to distinguish between a caspase-mediated intrinsic cell death program and an extracellular inflammatory component related to IL-1β.
Both IL-1β mRNA and caspase-1 expression are increased in the postischemic brain. While the increase in IL-1β mRNA occurs rapidly and can be attributed to endogenous cells in the brain, the increase in caspase-1 occurs in a delayed manner and is confined to activated microglia (Bhat et al., 1996). These changes in caspase-1 and IL-1β seem to have detrimental consequences in the ischemic brain because several studies have shown that a therapeutic benefit can be derived by blocking the action of IL-1β after an ischemic insult. Direct inhibition of IL-1β coupling to the IL-1β receptor clearly causes a reduction in infarct volume and cerebral edema in ischemia models (Betz et al., 1996) and the original observations have been validated in recent studies (Relton et al., 1996; Loddick and Rothwell, 1996b). Friedlander et al. (1996a) have described a transgenic mouse line that expresses an inhibitor of caspase-1 in neurons. These mice showed decreased infarcts and improved neurological outcome relative to their wild-type littermates. As a cautionary note, it should be remembered that the production of IL-1β during caspase-1 dependent apoptosis is not obligatory, although it can be a contributing factor (Friedlander et al., 1996b). It remains difficult to dissociate potentially apoptotic caspase-1 activation from IL-1β production in the ischemia models, and it is possible that some of the protective effects observed with caspase inhibitors involve anti-inflammatory effects rather than direct inhibition of apoptosis.
Caspase transcription in cerebral ischemia
Although it is important to recognize that transcription of new caspases is not a requirement for apoptosis because these enzymes are constitutively expressed as pro-enzymes and are activated by proteolysis, recent studies have examined the transcription of selected caspases after global and focal cerebral ischemia. After permanent focal ischemia, Nedd-2 and caspase-3 mRNA were induced between 8 and 16 hours after the insult, while the expression of caspase-1 remained constant (Asahi et al., 1997). These delays in expression are reasonably late in the ischemic cascade and their pathophysiological significance remains to be determined.
Caspase-1 mRNA is induced in the hippocampus after global ischemia (Bhat et al., 1996). The increased expression was observed 2 days after the transient ischemia and was localized to microglial cells. These data suggest the role of caspase-1 in global ischemia is linked to the activation of a local inflammatory cascade, rather than being a trigger for apoptosis.
p53 AND INDUCTION OF NEURONAL APOPTOSIS
One of the major dilemas in studies on gene expression induced by ischemia revolves around the significance of any findings. The overhanging question usually remains: is the observed change part of the injurious outcome, or part of an adaptive repair response to the initial ischemic insult (which may be protective during a second ischemic event)? This has been discussed with respect to immediate early genes (Akins et al., 1996; Herzog and Morgan, 1996) and heat shock proteins (Massa et al., 1996), but in reality should be considered for any observed change in gene expression after an ischemic episode. Studies on gene expression are largely still in the initial descriptive phase and a long way from an answer to this dilema. An additional caveat is the limitation of some studies to measurement of mRNA without demonstration of translation of the cognate functional protein.
The story of p53 and neuronal degeneration continues to evolve (Choi 1996; Chopp and Li 1996; Linnik 1996). This transcription factor sits at a crossroads between surveillance of the genome for DNA damage, recruitment of the repair machinery, or alternatively tipping of the cell into an apoptotic cell death pathway as a final solution in maintenance of tissue integrity (Levine 1997). Several additional experiments with p53-knockout-mice showing a less severe outcome in neurons after kainic acid treatment (Morrison et al., 1996) or adrenalectomy (Sakhi et al., 1996) add to those which have already shown attenuation of ischemic damage (Choi 1996; Chopp and Li 1996; Linnik 1996). Cultured neurons from such p53-deficient animals have a decreased sensitivity to either glutamate excitotoxicity or genotoxins (Enokido et al., 1996; Xiang et al., 1996). In addition, overexpression of p53 in either cultured cortical, hippocampal, or superior cervical ganglion neurons lead to apoptosis (Jordan et al., 1996; Slack et al., 1996; Xiang et al., 1996), while over-expression of a dominant-negative truncated p53 protected neurons from spontaneous apoptotic death (Eizenberg et al., 1996). Finally, increased expression of p53 has been reported after a cerebral ischemic insult (Chopp and Li, 1996, Tomasevic et al., 1996) although this is not always found (Nakagomi et al., 1996).
As a transcription factor, p53 upregulates several genes including bax and gadd45 (Smith and Fornace 1996; Levine 1997). Therefore it is of interest that, in this context, increases in bax have been reported to occur after cerebral ischemia in rats (Gillardon et al., 1996b; Chen et al., 1996), although in gerbils no increase in bax mRNA was seen (Honkaniemi et al., 1996a). Antisense oligonucleotides to bax prevented killing of cultured sympathetic neurons caused by neurotrophic deprivation (Gillardon et al. 1996c). gadd45 is also increased after focal (Jin et al., 1996) and global (Li et al., 1997) ischemia. While the function of Gadd45 is not fully elucidated, it has been shown to interact with PCNA which is a component of the DNA repair machinery (Kelman 1997; Smith and Fornace 1996). The selective depletion of PCNA in ischemia-sensitive CA1 cells of hippocampus is therefore highly suggestive that some failure of the restoration machinery is involved with selective neuronal death in this brain area (Tomasevic et al., 1996).
OXIDATIVE STRESS
What might induce p53 expression? It is clear that the large burst of oxidants produced during an ischemic reperfusion episode play a major role in the ensuing cerebral damage (Chan, 1996; Iadecola, 1997). Such reactive oxygen species are involved in neurotoxicity (Hosler and Brown, 1996) and can induce neuronal apoptosis (Bredesen 1996a, b ; McConkey and Orrenius, 1996; Mattson and Furukawa, 1996). The evidence for the importance of oxidative stress in ischemia comes from many directions including the use of pharmacological agents and antioxidants, and most compelling, from the use of transgenic and knockout animals for such genes as superoxide dismutase, catalase and nitric oxide synthase. A role for superoxide in producing ischemic neuronal death has been postulated in studies of transgenic mice overexpressing superoxide-dismutase (Chan 1996) but not in neuronal degeneration after kainate treatment (Kondo et al., 1997). Because reactive oxygen species such as hydroxyl, superoxide, or nitric oxide radicals are so transient and technically difficult to measure, direct evidence has been elusive. However, the report by P.K. Liu et al. (1996) contains direct measurements using a variety of analytical techniques and shows that after global cerebral ischemia, there was oxidative damage to the bases of DNA in the first hours of reperfusion, which although largely repaired, left some alterations which were also mutagenic. Because single strand breaks in neuronal DNA have also been described after ischemia (Tobita et al., 1995) or excitotoxicity in vitro (Didier et al., 1996), there is plenty of opportunity for discontinuities in the backbone of DNA to be a stimulant of the p53 surveillance system. A contrary report showing that, instead of oxidation leading to subsequent p53 activation, p53 itself acts downstream to regulate the intracellular redox state in smooth muscle is most provocative and should be examined in brain (Johnson et al., 1996).
Nitric oxide synthase
Besides hydroxyl and superoxide radicals, the nitric oxide (NO) radical is of great consequence in cerebral ischemia with its production increased at all stages subsequent to ischemic injury (Dawson and Dawson 1996; Iadecola, 1997; Szabo 1996). Nitric oxide is an intriguingly small cellular messenger acting as a neurotransmitter in both peripheral and central nervous systems, but also as a vasodilator. Nitric oxide is produced from L-arginine by three separate isoforms of the calcium calmodulin-dependent enzyme NO synthase (NOS) termed nNOS in neurons, and eNOS in cerebral endothelial and hippocampal pyramidal cells. Normally the third isoform, iNOS (immunological) is not present in the CNS, but iNOS becomes a major source of NO after infiltration of neutrophils caused by brain injury. The most apropos review for our present purpose is that of Iadecola (1997) which describes how the production of NO can be either beneficial (by eNOS) or detrimental (early by nNOS or later by iNOS). An increase in mRNA expression for both eNOS and nNOS occurs in the first hours after focal ischemia, to be followed up to 12 to 48 hours later by an increase in iNOS mRNA. The site of expression of the iNOS isoform can vary with whether the focal occlusion is transient or permanent: after transient occlusion of the middle cerebral artery, iNOS is expressed within 12 hours in endothelial cells, whereas after permanent occlusion iNOS is expressed after a delay of at least a day in infiltrating neurophils (Iadecola et al., 1996; Zhang et al., 1996). eNOS expression does increase in cerebral blood vessels after traumatic brain injury in which there is an ischemic component (Cobbs et al., 1997). Many pharmacological agents have been used to elucidate the role of NO after ischemia (eg Zhang et al., 1996), but the most striking exhibition of the detrimental action of NO has been shown by the attenuation of focal ischemic damage in mice with genetic knockout of either the nNOS or iNOS isoforms, but an opposite magnification of damage in mice with knockout of the eNOS isoform (Iadecola 1997). A decrease in hippocampal damage has also been shown in a global ischemia model in nNOS null mice (Panahian et al., 1996). The complexity of expression of NO coupled with the opposing actions of vasodilatory and excitotoxic actions make rational therapeutic targeting of NO non-trivial (Iadecola 1997). Besides discussion of NO, the importance of combination of NO with superoxide to form extremely noxious peroxynitrite has been addressed (Dawson and Dawson 1996; Szabo 1996). Peroxynitrite can cause DNA strand breaks, stimulate poly(ADP-ribosyl) polymerase and subsequently induce neuronal apoptosis. Using cultured cortical cells from nNOS null mice, it has been shown that NO-mediated neurotoxicity is primarily associated with activation of NMDA (and not kainate) receptors, and the provocative statement made that “NO may be responsible for as much as 80% of NMDA neurotoxicity (Dawson et al., 1996).
Mitochondria and apoptosis
A heuristic body of work has emerged from attempts to build a cell-free system that would recapitulate the major nuclear events of cell death (Kroemer et al., 1997). Whether induced by oxidative stress or other stimuli, alterations in mitochondrial function accompany apoptosis. There is a precipitous drop in mitochondrial membrane potential that precedes formation of superoxide and DNA fragmentation (Kroemer et al., 1997; McConkey and Orrenius 1996). This transmembrane potential exists because of the assymetric distribution of protons on either side of the inner mitochondrial membrane resulting in an electrical and protonic gradient that is essential for mitochondrial function. The collapse of these gradients in apoptotic cells (including dying neurons) may be caused directly by opening of the permeability transition pore whose composition is unknown but which contains multimeric protein complexes. In building cell-free systems to study cell death, it can be shown that cytoplasmic extracts of apoptotic cells can induce normal nuclei to show morphological characteristics and cleavage profiles of both protein and DNA indicative of apoptosis (X. Liu et al., 1996). In some of these cell-free systems, the cytoplasmic extracts have to be enriched with mitochondria (Zamzami et al., 1996). How failing mitochondria with open pores may directly induce apoptosis has possibly been revealed by the demonstration of release of proteins from mitochondria, for example cytochrome C (X Liu et al., 1996; Yang et al., 1997) or a 50-kD apoptosis-inducing factor (Susin et al., 1996), both of which are involved in some way with the activation of caspases (Kluck et al., 1997; Susin et al., 1996; Yang et al., 1997). The mitochondrial factors responsible for induction of apoptotic nuclear changes are not encoded by mtDNA (Marchetti et al., 1996). The exact sequence of events may be different for the different inducers. In the Xenopus egg or mammalian cell models the release of cytochrome C precedes any collapse of mitochondrial gradients or permeability transition (Yang et al., 1997; Kluck et al., 1997), whereas in mammalian cells a mitochondrial pore opening comes before the release of the 50 kD apoptogenic protein (Susin et al., 1996; Zamzami et al., 1996). A critical sensor of the redox state of cells which links oxidative stress to mitochondria to apoptogenic protein release may reside in mitochondrial thiol groups (Marchetti et al., 1997).
Mitochondrial deenergization occurs in cultured neurons induced to die by glutamate (Ankarcrona et al., 1996a), and has long been known to be associated with neuronal cell death after episodes of cerebral ischemia (Kristian and Siesjo 1996; Siesjo and Siesjo 1996). This deenergization is associated with collapse of the mitochondrial membrane potential and is accompanied by a permeability transition and the disruption of calcium homeostasis. Cyclosporin A has the capacity to prevent the permeability transition (Kroemer et al., 1997), and it is therefore of significance to our discussion that this immunosuppressor can significantly reduce ischemic damage (Siesjo and Siesjo 1996) and block glutamate toxicity in vitro (Ankarcrona et al., 1996a). Also of significance is that the antiapoptotic gene Bcl-2 can prevent the mitochondrial release of both the 50 kD apoptogenic protein (Susin et al., 1996) and of cytochrome C (Yang et al., 1997; Kluck et al., 1997). The continued application of these ideas in the arena of neuronal cells is greatly anticipated.
DNA FRAGMENTATION IN ISCHEMIC BRAIN
Because oligonucleosomal DNA fragmentation is a major hallmark of apoptosis (Bredesen 1996b; Hale et al., 1996; Vaux and Strasser, 1996), it is no wonder that it has been widely applied in investigations to ascertain a role for apoptosis in neuronal death. There have been innumerable reports showing electrophoretic patterns of laddered DNA fragments from either ischemic brain or other neurodegenerative situations (Charriaut-Merlangue et al., 1996a; Choi, 1996; Chopp and Li, 1996; Mehmet and Edwards, 1996). Besides these 200 to 1000 bp low molecular weight DNA fragments, higher order fragments of 10, 50, or 300 to 700 kbp have also been detected after focal (Charriault-Merlangue et al., 1996a) or global (MacManus et al., 1997) ischemic insults, and also following glutamate toxicity in cultured cerebellar granule cells (Ankarcrona et al., 1996b).
The use of in situ end-labelling (TUNEL with terminal transferase or ISEL with polymerase) to detect DNA fragmentation in brain slices has also been extensive. Separately or in combination, these two techniques continue to be used in studies after either transient global (MacManus et al., 1997; Schmidt-Kastner et al., 1997) or focal ischemia in adult animals (Charriault-Merlangue et al., 1996b; Du et al., 1996; Gillardon et al., 1996a, Gillardon et al., 1996b; Jin et al., 1996; States et al., 1996), neonatal hypoxia-ischemia (Walton et al., 1996b) or excitotoxicity (Kondo et al., 1997; Portera-Calliau et al., 1997a). These methods have also been used successfully to show granule cell death after cortical impact injury (Colicos and Dash, 1996), thalamic neuronal death after thiamine deficiency (Matsushima et al., 1997), or abbrogation of neuronal degeneration in p53 knockout mice in response to kainic acid or adrenalectomy (Morrison et al., 1996; Sakhi et al., 1996). The comparison of permanent with transient middle cerebral artery occlusion in mice indicated that less in situ end-labelling occurred in the absence of reperfusion (Murakami et al., 1997). A failure to show DNA fragmentation by these methods has been noted in prolonged somal survival of septohippocampal neurons after fornix-fimbria transection (Butterworth and Dragunow, 1996), but the presence of TUNEL-positive cells without the convergent demonstration of electrophoretic demonstrable DNA fragmentation after kainic acid induced seizures has been reported (Weiss et al., 1996).
Apoptotic endonucleases
Most of the studies cited above that show oligonucleosomal DNA fragmentation after cerebral ischemia go no further with the analysis. It is concluded that the fragmentation is apoptotic. However, it may not be so simple. In a study that examined the end-groups produced in high molecular weight (10 to 50 kbp) DNA fragments in ischemic brain it was shown that, besides the production of 3'-OH end-groups which are documented in apoptosis, 5'-OH end-groups were also detectable (MacManus et al., 1997). It was argued that at least two endonucleases would be needed to produce such fragmentation, and perhaps a third endonuclease to complete cleavage to the widely demonstrated oligonucleosomal fragmentation. What is the nature of the endonuclease producing DNA fragmentation in ischemic brain? This remains unknown because there are no published reports on specific brain enzymes, but there are many candidates. Besides several endonucleases such as DNAse I and II and Nuc18 which have been implicated in apoptosis in non-neuronal cells (Bortner et al., 1995; Walker et al., 1995), additional potential endonucleases have been described in apoptotic hepatoma (Pandey et al., 1997), lymphocytes (Collins et al., 1996; Khodarev and Ashwell, 1996), and human leukemic cells (Fraser et al., 1996). It remains to be seen if any of these have a role to play in ischemic neuronal DNA fragmentation. The report of Collins et al. (1996) is of particular interest because the newly described endonuclease produces DNA cleavage with 5'-OH end-groups in a DNAse II manner, although with a molecular weight at 45 kD it cannot be DNAse II (33 kD). Another issue relevant to ischemic cell death is the acidification during apoptotic cell death (McConkey and Orrenius 1996) which may activate acidic endonucleases such as DNAse II (Bortner et al., 1995; Walker et al., 1995). In addition to DNAse, II several other acidic endonucleases have recently been described (Collins et al., 1996; Gottlieb et al., 1996; Ostad et al., 1996). The early intracellular acidification before detectable DNA fragmentation has been documented by flow cytometry by Gottlieb et al. (1996). Also of interest is that the simple lowering of the pH in the culture medium of HL60 cells from pH 7.6 to pH 6.8 was sufficient to induce apoptotic DNA fragmentation (Park et al., 1996), and the intriguing idea was raised that oncogene blockage of apoptosis may be because o a prevention of acidification (Ostad et al., 1996). Because of the long-established acidification of the ischemic brain caused by lactate accumulation, all of the work on cellular acidification and activation of acid endonuclease in standard cancer cell lines needs to be revisited in a neuronal context.
The hunt for the elusive endonuclease responsible for apoptotic oligonucleosomal DNA fragmentation has not bagged any prize enzyme, which is irrefutably the one which gives the observed electroporetic patterns. An alternate, and sobering, idea is that this hunt is futile. Provocative experiments suggest that the cleavage of DNA may not be enzymatic, but produced instead by chemical nuclease action, most likely by hydroxyl radical. A system of hydroxyl radical generation using copper phenanthroline complexes were shown to be capable of generating laddered DNA fragments in isolated liver nuclei (Burkitt et al. 1996) and also in cultured liver cells (Tsang et al. 1996). Similar findings with isolated chromatin (presumably lacking any contaminating endonuclease) suggest some preferential modification of internucleosomal regions perhaps by the insertion of 8-oxoG lesions in the DNA backbone (Enright et al. 1996; Tsang et al. 1996) which would lead to subsequent strand scission. This somewhat heretical suggestion of chemical nuclease action by hydroxyl radical cannot be overlooked in the area of cerebral ischemia because of the evidence, alluded to above, of oxidative damage to DNA after global ischemia (Chan 1996; PK Liu et al. 1996). In this regard, the family of repair endonucleases (apurinic endonucleases) involved with base-excision repair of DNA and which are capable of recognizing some of the oxidative base lesions described in ischemic brain (PK Liu et al., 1996) may become involved. These repair endonucleases are expressed in brain (Wilson et al., 1996), but the single report concerning ischemia indicates a reduction of this enzyme in CA1 hippocampal cells before DNA fragmentation (Walton et al., 1996a). Because the majority of DNA damage produced after an ischemic insult is rapidly repaired (P. K. Liu et al., 1996), other repair endonucleolytic activities such as those involved with nucleotide-excision repair must be responsible. Another cellular entity responding to radical induced DNA damage is poly(ADP-ribosyl) polymerase (Szabo 1996), and the possibility should be considered that the increased susceptibility of chromatin to cellular endonuclease may also be achieved by poly(ADP-ribosyl)ation of histone H1, which resides in the internucleosomal linker region (Yoon et al., 1996).
APOPTOTIC MORPHOLOGY AFTER CEREBRAL ISCHEMIA: A VEXATIOUS ISSUE
Distinguishing apoptosis from necrosis can be a contentious task (Choi 1996). Apoptosis was first delineated from necrosis in 1972 on morphological grounds using electron microscopy. In neurons, as in other cells, the principal observable feature in the light microscope is nuclear “aggregation of chromatin into dense sharply delineated masses” (Charriault-Marlangue et al., 1996a). There also has been wide use of in situ end-labelling to detect DNA fragmentation (see above) which unfortunately alone cannot discriminate between apoptosis and necrosis although occasionally claims to the contrary are made based on intensity and location of label (Charriault-Marlangue et al., 1996a). Although some scepticism exists (Yanker 1996), this lack of discrimination is not appreciated by many and has lead to the deplorable use of TUNEL as a facile yardstick for the presence of apoptotic neurons in a whole host of diseases such as Alzheimer's and Parkinson's. It is to be hoped that further work will prove these hasty conclusions to be correct.
Neurons dying by programmed cell death during development do show morphological (and biochemical) hallmarks of apoptosis (Portera-Calliau et al., 1997a; Ross 1996). In neonatal models of hypoxia/ischemia, the dying neurons show chromatin coalescence (karyor-rhexis) and laddered DNA fragmentation (Edwards and Mehmet 1996; Ferrer 1996), and have an neonatal age-dependent exhibition of apoptotic morphology in response to excitotoxicity (Kelly and Burke, 1996; Portera-Calliau et al., 1997a). However, in adult models of ischemia, the morphological evidence to date is not consistent to say the least (Charriault-Marlangue et al., 1996a; Kirino 1996). Coalescence of chromatin has been recorded after focal ischemia particularly in the penumbral area on the edge of the infarct (Charriault-Marlangue et al., 1996b; Chopp and Li 1996; States et al., 1996), but this was not confirmed in a rigorous ultrastructural study (van Lookeren Campagne and Gill, 1996). Also to be considered, albeit only with light microscopic resolution, is the lack of coalesced or marginated nuclear morphology in TUNEL or ISEL end-labeled nuclei in myriad previous reports on adult ischemic animals, and including those recently appearing (Du et al., 1996; Gillardon et al., 1996a, Gillardon et al., 1996b; Jin et al, 1996; Schmidt-Kastner et al., 1997). It is always appropriate in times of doubt to revisit the old masters. A quote from one of the founding fathers of the field of apoptosis should give pause: “The ultrastructural confirmation of light microscopic identification of dying cells will be evident. Despite the difference between the processes of apoptosis and necrosis, the terms pyknosis and karyor-rhexis can be legitimately used to describe the appearances in the light microscope of certain phases in the evolution of both” (Wyllie et al., 1980).
Not apoptosis nor necrosis
Instead of trying to apply black and white definitions of apoptosis versus necrosis to ischemic cell death based on the inadequate light microscopy evidence, a more useful postulate may be that neuronal cell death after ischemia should be considered in its own right without name calling. Maybe those using other models of neuronal degeneration have already pointed the way in showing that, for example, cortical impact injury elicits hippocampal damage (Colicos and Dash 1996), or neonatal excitotoxicity elicits cortical damage (Portera-Calliau et al., 1997a), with features of both apoptosis (nuclear margination) and necrosis (mitochondrial swelling) in the same cells. Another angle was presented in the above cited exemplary study of excitotoxicity using both neonatal (Portera-Calliau et al., 1997a) and adult (Portera-Calliau et al., 1997b) models, with the most elegant exposition of a morphological continuum with classical apoptosis and necrosis at either extreme. The secondary necrosis noted may be the normal end-stage of the apoptotic process in neurons which die, for example after ischemia, in such large numbers that the phagocytotic process cannot clear them efficiently. An added intriguing intricacy in adult excitotoxicity is the finding that, independent of agonist concentration, NMDA and non-NMDA agonists produce different neuronal morphologies that resemble necrosis and apoptosis respectively but share similar laddered DNA fragmentation (Portera-Calliau et al., 1997b). Finally, perhaps the type of cell death is determined postsynaptically because repetitive perforant path stimulation can lead to either apoptosis (granule cells) or necrosis (pyramidal cells) in hippocampal neurons (Sloviter et al., 1996).
IMMEDIATE EARLY GENES AND HEAT SHOCK PROTEINS
Reams of data have been accumulated in this decade on the rapid but transient induction of immediate early genes (IEG) after various stimuli including either a focal or global ischemic insult (Akins et al., 1996; Herzog and Morgan 1996; Hughes and Dragunow 1995). These transcription factors, exemplified by the leucine-zipper proteins Fos and Jun, are considered to be nuclear third messengers that link extracellular signals to longer term alterations in effector gene expression, and to be a paradigm for transcription-dependent i.e., programmed cell death (apoptosis) even in a situation of overall defective protein synthesis such as ischemia (Akins et al., 1996; Herzog and Morgan 1996; Wiessner et al., 1996). In the normal CNS, these proteins are present at relatively low levels but correlations have been made between increased expression of fos and jun and subsequent delayed neuronal death induced by ischemia or a variety of disease states such as epilepsy. A causal link of IEG expression to subsequent cell death was made in cultured neurons using antibodies or dominant-negative Jun that prevented the ultimate demise of treated cells (Herzog and Morgan 1996). The intense research activity in this area has cooled in recent years but several reports continue to argue the relevance of increases in IEG mRNA after global and focal ischemia in the adult (Gillardon et al., 1996b; Lin et al., 1997; Nakagomi et al., 1996; Wiessner et al., 1996) and neonate (Ferrer 1996), or after experimental brain injury (Raghupathi and McIntosh 1996). Another family of IEG are the zinc-finger family, which include NGFI-A, B and C, egr-2 and 3, and Nurrl, and have been shown to change after global ischemia (Akins et al., 1996; Honkaniemi and Sharp 1996b). All of these transcription factors (except egr-2 and Nurr1) were induced in brain areas, especially hippocampus, which are known to be susceptible to ischemic damage. Persistent expression of these IEG did not correlate with whether cells are destined to die because NGFI-C and egr-3 were present in dentate granule neurons for a prolonged period. It was suggested that the combination of transcriptional activation with lack of negative feedback because of blocked translation might explain the general prolonged expression of these IEG in particular, but also perhaps in general to include Fos and Jun (Honkaniemi and Sharp 1996b).
Immediate early genes and repair
Despite the above apparently irrefutable evidence for a role in cell death for the IEG, an disconcerting alternate view suggests that fos and jun may be components of an adaptive or repair/regeneration process because many cells that have increased expression actually survive insults such as excitotoxicity (Akins et al., 1996; Herzog and Morgan 1996; Hughes and Dragunow 1995). Such a radical idea also comes from the surprising finding of normal cell death in fos-null mice, and the lack of effect on neuronal cell death in the spinal chord after sciatic nerve resection in such knockout animals (Roffler-Tarlov et al., 1996). The clever use of fasting as a challenge to such a nuturing role for c-fos yielded corroboration in the demonstration of prolonged and enhanced expression after focal ischemia in hypoglycemic animals which are well known to have less damage following an ischemic insult (Lin et al., 1997). Further support comes from studies showing increased fos or junD mRNA in the resistant hippocampal dentate area using a chest compression model of global cerebral ischemia (Tseng et al., 1997), or after transient global ischemia with postischemic hypothermia (Kamme and Wieloch 1996), respectively. A third view would be that the IEG are involved in both cell death and adaptive/stress response, and it is the cellular context that is important, eg hippocampal CA1 pyramidal or dentate granule cells. It is this context that is not understood (McConkey and Orrenius 1996). The statement made at the beginning of this article could be rewritten to “signals that promote apoptosis in one cell type can suppress cell death in another”.
Heat shock proteins
Another aspect of an adaptive/regeneration response in the brain to ischemic insults concerns heat shock proteins whose expression is increased in both neurons and glial cells (Akins et al., 1996; Kirino 1996; Massa et al., 1996). Global ischemia induced hsp70 expression throughout the hippocampal area before any degeneration of the CA1 layer (Wiessner et al., 1996), and after focal ischemia, cells that had increased expression of hsp70 did not show DNA fragmentation by in situ end-labelling (States et al., 1996). From such studies arise ideas that the hsp70-stained cells are reversibly injured and likely survive the ischemic insult, perhaps in a way analogous to the cells identified above as gadd45 positive (Jin et al., 1996), or c-fos positive (Lin et al., 1997). Because an ischemic insult does engender a stress response, it should not be a surprise that accumulating evidence indicates that brief, not obviously harmful, episodes of ischemia in either brain or heart induce tolerance to a second ischemic insult (Chen and Simon 1997; Massa et al., 1996). In either case of induced tolerance, increased cerebral gene expression would seem to be involved, and attractive candidates would be IEG, heat shock proteins such as hsp70, or antiapoptotic gene families such as the Bcl-2 family or the IAP family (Liston et al., 1996).
MINORITY REPORTS: Myc, NEURONAL APOPTOSIS INHIBITORY PROTEIN, HYPOXIA-INDUCED GENES, AND TRANSCRIPTION REGULATION
The preceeding text has been governed by the demographics of the extant literature on gene expression, primarily in neurons, after an ischemic insult to the brain. However, research in other areas must not be ignored. There have been individual reports of interest which may blossom in the future to the monumental impact of the IEG, bcl2 or caspases, and extend studies beyond the neurocentrism of the moment.
Myc
Besides fos, jun and p53, cancer research has also led to the recognition of the importance of another transcription factor, c-Myc, in regulating apoptosis (Henriksson and Luscher 1996; Zorning and Evan 1996). The nuclear phosphoprotein c-Myc is involved in cell cycle regulation and is not generally expressed in nonproliferating or quiescent cells. Few genes have actually been identified as targets for c-Myc transactivation, but one is p53. Overexpression of c-myc primes cells for apoptosis and facilitates the adverse effects of lack of nutrients or oxygen (Alarcon et al. 1996; Graeber et al., 1996). Such conditions exist in the ischemic brain and it is therefore of interest that an up-regulation of c-myc expression has been reported after focal occlusion (Nakagomi et al., 1996). Another target gene of c-Myc is ornithine decarboxylase (Henriksson and Luscher 1996) which has been seen to increase after cerebral ischemia, although its overexpression in transgenic rats was unsuccessful in influencing infarct volume (Lukkarinen et al., 1997).
Neuronal apoptosis inhibitory protein
Besides Bcl2, there is a another family of proteins known to inhibit apoptosis: the inhibitors of apoptosis family, which was first identified in viruses such as baculovirus or cowpox virus that impede apoptosis to prolong the life of infected host cells (Bredesen 1996b;Vaux and Strasser 1996). The mammalian homologues have now been identified and shown to inhibit apoptosis in mammalian models (Duckett et al., 1996; Liston et al., 1996; Uren et al., 1996). One of these homologues is the gene for spinal muscular atrophy (neuronal apoptosis inhibitory protein) which is widely expressed in the brain, but in particularly high levels in exactly those neuronal populations that are known to degenerate in diseased children (Xu et al., 1997). Studies on the expression of these antiapoptotic genes in other neuronally injurious situations including cerebral ischemia is keenly awaited.
Hypoxia-regulated genes
The expression of a novel oxygen regulated stress protein ORP150 was described in ischemic mouse brain (Kuwabara et al., 1996), and may be the first signs of a research thrust in the area concerning genes of the brain that are regulated by hypoxia. Cancer research is again heuristic in demonstrating that tissues have a plethora of responses available when confronted with hypoxia, including upregulation of many transcription factors such as fos, jun, and p53, in addition to stress proteins such as hsp70. Another helix-loop-helix transcription factor first described in 1993, hypoxia-inducible factor, has been shown to influence the expression of many oxygen-regulated genes (Dachs and Stratford, 1996). The recognized target repertoire of hypoxia-inducible factor-1 now includes genes such as erythropoietin, vascular endothelial growth factor, aldolase, enolase, lactate dehydrogenase, and heme oxygenase-1 (Lee et al., 1997; Semenza et al., 1996). Hypoxia-inducible factor-1 is present in brain, and its expression increases after exposure of animals to hypoxia (Wiener et al., 1996). Therefore, increased gene expression for heme-oxygenase after either global or focal ischemia may come as no surprise (Massa et al., 1996; Geddes et al., 1996; Takeda et al., 1996). It is easily predicted that this arena will become more crowded in the coming few years as the potential for exploiting new knowledge in oxygen-controlled gene expression in the brain is realized.
Transcription regulation
Many of the stroke-induced changes in gene expression result from changes in the relative distribution of the transcription factors that transactivate these genes. Some changes in transcription factor expression can be directly linked to cell death, such as the induction of the transcription factor p53 transactivating the proapoptotic protein Bax (Smith and Fornace, 1996; Levine, 1997). However, interpreting the consequences of changes in the expression of transcription factors requires careful analysis because these factors are highly context dependent. A good example of this context dependency is NF-κB, a transcription factor that is sequestered in the cytoplasm by the inhibitory protein IκB (Lipton, 1997; and Baichwal, and Baeuerle 1997). When IκB is phosphorylated and/or cleaved, NF-κB is free to translocate to the nucleus where it influences the expression of a wide variety of genes, including many stress activated genes. The levels of NF-κB are elevated in several degenerative diseases, including stroke (Terai et al., 1996a) and Alzheimer's disease (Kaltschmidt et al., 1997; Terai et al., 1996b; Barger and Mattson 1996) and Grilli et al. (1996) showed that blocking NF-κB activation by aspirin or sodium salicylate could protect neurons against an excitotoxic insult. While these data suggest that NF-κB activation is detrimental, several studies have shown that NF-κB activation blocks apoptosis to a variety of insults (Beg and Baltimore, 1996; Wang et al., 1996; Van Antwerp et al., 1996; ZG Liu et al., 1996) and can be neuroprotective (Goodman and Mattson 1996). Thus, this well understood transcription factor can be both protective and detrimental, depending on the context in which it is expressed and the nature of the insult. Transcriptional regulation remain relatively unexplored in the context of ischemic neuronal death, and these types of studies will be necessary to fully understand the basis for changes in gene expression that are induced by cerebral ischemia.
Minorities
There are scattered reports of increases in neurotrophins and various excitatory amino acid receptors following an ischemic insult which have been reviewed by Akins et al. (1996). The link between cell proliferation and apoptosis has been touched on by many (Desbarats et al., 1996; McConkey and Orrenius (1996) extended to neurgenesis and neuronal apoptosis as well (Ross 1996). After kainic acid treatment, an increase of the cell-cycle protein cyclin D1 was reported in astrocytes (W Liu et al., 1996). Ischemia-induced increases in gene expression in astrocytes were described for β-amyloid precursor proteins (Kositinaho et al., 1996), clusterin (Walton et al., 1996b), and the neurofibromatosis gene product NF1 (Giordano et al., 1996). What function these may have is unclear, but these upregulated gene products may be involved in the response to injury and the initiation of cellular remodelling. There have been singularly few studies on ischemic human brain but a notable exception showed increases in TGF-β1 in all cell types, particularly in the penumbral zone (Krupinski et al., 1996).
CONCLUSION
It is hoped that the basic science in animal models will be applicable to the human condition of stroke. Encouragement for continuation with investigation in fundamental mechanisms of cell death come from the finding of substantial amounts of potentially viable brain tissue in stroke patients (Marchal et al., 1996).
The early work on IEG after ischemia was inspirational and indubitably encouraged further work on expression of other genes as discussed in this article. The realization that, despite a decade of work by a host of investigators producing a mountain of publications, the last word on IEG in cell death is a long way from being written is a sobering thought. Much patience and dedicated research lie ahead before as much wisdom as has been acquired from IEG research will be available for the host of other genes under investigation after an ischemic insult. The basic preclinical research on this subject continues apace (Karpati et al., 1996) with the hope of defining practical concepts that can be implemented in the clinic.
Footnotes
Abbreviations used
Acknowledgements
The authors thank our many colleagues who helped to make this review comprehensive by providing insight and copies of publications before publication.