
Abstract
Implementation of regenerative medicine in the clinical setting requires not only biological inventions, but also the development of reproducible and safe method for cell isolation and expansion. As the currently used manual techniques do not fulfill these requirements, there is a clear need to develop an adequate robotic platform for automated, large-scale production of cells or cell-based products. Here, we demonstrate an automated liquid-handling cell-culture platform that can be used to isolate, expand, and characterize human primary cells (e.g., from intervertebral disc tissue) with results that are comparable to the manual procedure. Specifically, no differences could be observed for cell yield, viability, aggregation rate, growth rate, and phenotype. Importantly, all steps-from the enzymatic isolation of cells through the biopsy to the final quality control-can be performed completely by the automated system because of novel tools that were incorporated into the platform. This automated cell-culture platform can therefore replace entirely manual processes in areas that require high throughput while maintaining stability and safety, such as clinical or industrial settings.
Keywords
Introduction
Regenerative medicine, which combines recent core disciplines such as tissue engineering, biochemistry, and applied bioengineering, is one of the most promising yet at the same time challenging research fields. The overall goal is to develop or produce cells or cell-based products to regenerate or repair a wide range of damaged or degenerated tissues or organs. Although our knowledge of regenerative medicine has significantly increased and tremendous research funds have been spent since the 1980s, there are only few products currently available on the market, mostly in the areas of skin replacement and cartilage substitutes (co.don, Tetlow, Germany; apligraft, Organogenesis, Canton, MA; and dermagraft, Advanced Biohealing, LaJolla, CA). This is mostly because too little effort has been made so far to turn small-scale laboratory developments into clinical products that are safe, reproducible, and cost efficient, thus possessing sufficient potential for a commercial application.
Manual standard procedures currently used for the isolation and culture of human primary cells, which are most likely to be used in regenerative medicine, are labor intensive and thus cost intensive while possessing undesirable operator-dependent variation. Primary cells are usually isolated from the bare minimum of a biopsy, resulting in a very low initial cell number, especially if tissues with low cellularity 1 are used. Therefore, serial passaging is needed to obtain the high cell numbers required for clinical applications. Depending on the proliferation potential of the respective cell type, this in vitro expansion can require not only days, but even weeks, thus increasing costs and inconsistency in handling and, in the end, differences in the final product. Automation of cell isolation, expansion, and characterization will thus be essential to improve safety, reproducibility, quality control, and scalability. 2,3
Several academic groups, often in collaboration with an industry partner, have tried to develop automated cell-culture platforms in the last few years, 4 –10 mostly using different types of mesenchymal stem cells. Although stem cells are obviously an interesting and promising cell type for regenerative medicine because of unique properties such as their differentiation potential, they are on the other hand fast proliferating compared with many other cell types. It is therefore essential to test automated systems also with primary cells, which already pose a challenge when using manual procedures, but involve fewer safety concerns than stem cells.
In the present study, human intervertebral disc cells were chosen because of their clinical relevance and specific cellular properties. Intervertebral disc degeneration is an area with a high clinical demand because of a lack of reliable therapies and high health-care costs. 11 In 2005, Americans with back pain caused estimated total costs of $85.9 billion, not including indirect costs because of sick leave and other associated consequences. 12 Back pain can have multiple sources, one being degeneration of the intervertebral disc, a major load bearing structure in the spine. Disc degeneration starts early in life because of a very limited nutrition of the tissue based on its avascularity. 13 However, treatment options are rare and inefficient. Therefore, cell therapy could be a promising new approach, but culture of disc cells has proven to be very challenging: Not only is the cell number in vivo very low, 1 but additionally, cells show poor proliferation and metabolic activity 14 while being prone to dedifferentiation in prolonged 2D expansion, 15,16 similar to chondrocytes. Increasing the low metabolic activity of disc cells (especially when obtained from degenerated tissue) by stimulating matrix production (e.g., via growth factor treatment) may be necessary for sufficient tissue regeneration. Alternatively, mesenchymal stem cells from easily accessible sources (e.g., fat) could possibly be used for intervertebral disc regeneration as well, although initiation of specific differentiation is not yet established.
In this article, we describe an innovative automated system for the isolation, expansion, and characterization of primary cells that is basedon aliquid-handling robot from Tecan (Switzerland), integrated with modules for sterile operation, tissue manipulation and homogenization, cell growth, and pheno-type analysis. Performance of the automated system is compared with standard manual cell-culture techniques with regard to cell yield, viability, aggregation rate, growth rate, and phenotype, using human intervertebral disc cells as a model cell type.
Materials and Methods
Materials
Biopsies. Human intervertebral disc tissue was excised from patients undergoing spinal surgery (disc herniation or degeneration) with a mean age of 49.4 years for experiments on cell isolation (n = 5, SD = 18.5) (Fig. 1).
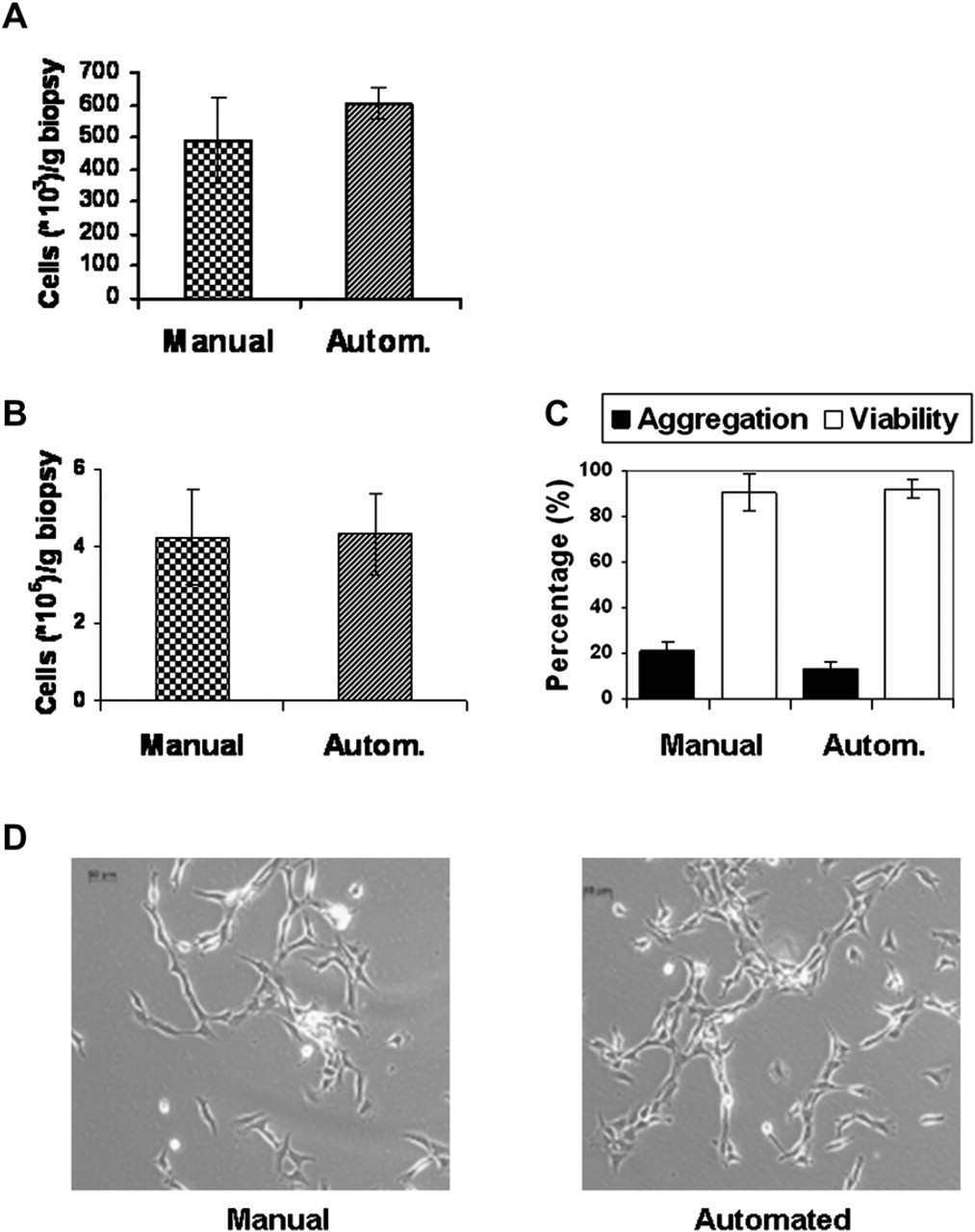
Cells obtained after isolation. A. Cells are counted at the end of the manual or automated (autom.) isolation process. Measurement means
Instrumentation
The automated cell-culture platform (Fig. 2) is based on the liquid-handling robot Freedom EVO 150 (Tecan, Switzerland) and was developed to suit cell-culture requirements by adding the following modules:
Liquid-handling arm with four standard tips and four wide bore tips for cell pipetting
Controlled pump option for accurate dispensing of media.
10-Way valve for selecting media or other liquids
Robotic manipulator arm (RoMa) equipped with eccentric gripper fingers
Orbital shakers (2×)
Flipper module with four positions for holding RoboFlasks (Corning, NY)
Centrifuge Rotanta 46 RSC Robotic (Hettich, Germany), mounted under the worktable (which can be loaded and unloaded by RoMa)
Clean air hood on top of the system causing vertical, low turbulence displacement flow (0.25 m/s ± 20%) to obtain purity level four requirements (VDI 2083) and class 1000 (US Federal-Standard 209)
Dispomix (Medic Tool, Switzerland), a programmable homogenizer for tissue desegregation in closed tube
Cellavista (Roche, Switzerland), an automated microscopic cell analyzer platform for noninvasive brightfield and fluorescence applications
Dedicated incubator (37 °C, 5% CO2, humidified atmosphere) (can be added)

The automation platform. The automation platform based on Freedom EVO (Tecan, Switzerland) 150 includes: Clean air cabinet (1), Robotic manipulator arm for moving automation-friendly cell-culture flasks (RoboFlask, Corning) (2), Liquid-handling arm with steel tips (3), Image-based cellular analyser (Cellavista, Roche) (4), Flask flipper module (5), Robotic shaker (6), Centrifuge (7), and Cell incubator (not shown).
The pipetting software Freedom EVOware (Tecan, Switzerland) groups pipetting commands and device commands into scripts and allows creation of user- and cell-type-specific experimental programs. RoboFlasks can be automatically loaded and unloaded using the RoMa.
Tissue Preparation and Cell Isolation
Human intervertebral disc biopsies (n = 5) (0.4–6.5 g) were excised during spinal surgery at an Orthopedic University Hospital from patients suffering from disc degeneration or herniation. The approval of the local ethics committee and informed consent from the patients was obtained. The biopsy was stored in DMEM/F12 with 1% Penicillin, Streptomycin, Amphotericin and processed within 4 h after surgery.
For the manual isolation, biopsies were rinsed with phosphate buffered saline (PBS), cut into small fragments, transferred in a falcon tube, and digested by 15 mL of 0.2 U/mL collagenase NB4 in DMEM/F12 with 1% antibiotics/anti-mycotics for 16 h at 37 °C and 5% CO2. Digestion was stopped by addition of DMEM/F12 with 5% FCS and 1% antibiotics/antimycotics. The cell pellet (obtained by centrifugation at 200 g for 5 min) was washed twice with PBS, resuspended in proliferation medium (5% FCS, 1% Penicillin-Streptomycin, 5 ng/mL FGF-α, 1 ng/mL TGF-β in DMEM/F12) and transferred into a RoboFlask at a cell density of 2,000 cells/cm2 (cell counting using hemocytometer).
For the automated isolation, the biopsies were rinsed with PBS and transferred into a Dispomix tube with 15 mL collagenase solution (see above). Tissue dissociation was performed by two cycles of homogenization (0–700 rpm for 1 min, 0–3,000 rpm for 15 s) and digestion as described for the manual procedure. The cell suspension was washed twice by the automated system, resuspended by vortexing, liquid aspiration, and dispension by the robot and transferred into a RoboFlask at a cell density of 2,000 cells/cm2 (cell amount determined using an automated cell counter, Cedex, Roche, Switzerland).
Cell Expansion
For cell expansion, the medium was changed twice a week, either manually or with the automated system.
When a confluent state was reached, determined in the manual process via microscopic evaluation and in the automated process with the Cellavista analyzer, cells were harvested and reseeded at a density of 2,000 cells/cm2 (for details see Table 1).
Comparison of manual and automated procedure for splitting of cells

Quality Control in the Automated System
Immunofluorescent analysis was performed using the Cellavista analyzer with an appropriate fluorescent filter set (automated process) or mounted and visualized using an Axioscope 2 Plus microscope (Zeiss, Switzerland) (manual process).
Statistical Analysis
Results are expressed as mean ± standard deviation (SD) from independent experiments (sample numbers as shown in results). Quantitative data was analyzed with an unpaired, two-tailed Student's t test, with a significance level of p < 0.05.
Results
Sterility
Using a closed liquid system and regular wash procedures (pumping 0.4% perchloric acid in the tubing system followed by rinsing with sterile deionized water) enabled creation of system sterility with no microbial contamination even after prolonged culture in antibiotic-free medium for at least 21 days (data not shown).
Cellular Characteristics Directly after Isolation
Manual mincing of the biopsy using a scalpel blade (2–3–mm pieces) and automated mincing using the Dispomix tool, followed by digestion in enzyme solution for 16 h in both systems produced a comparable result (n = 5), with a trend toward higher values for the automated system (manual = 478 ± 133 × 103 cells/g; automated = 607 ± 52 × 103 cells/g) (Fig. 1A). The automated procedure showed statistically significant (p = 0.043) higher reproducibility as indicated by the lower SDs (manual = 27.8% of mean; automated = 8.5% of mean).
Cellular Characteristics during Culture
In both systems, cell amount was similar (about 4.1 × 106 cells/g biopsy) (Fig. 1A, 7 days), cell viability was >90% (Fig. 1B, 7 days), and the aggregation rate was 15–20% (Fig. 1B, 7 days), without any differences in cell morphology (Fig. 1D, 4 days) (n = 5).
During prolonged culture, cells were passaged (see Table 1), with seeding densities of 2,000 cells/cm2. In passage 2, cell counts at 70% confluence (microscopic observation) yielded approximately 8.5 × 106 cells/RoboFlask (Fig. 3A), with >99% viability (Fig. 3B) and approximately 15% aggregation rate (Fig. 3C) in both systems. No statistically significant differences could be observed for any parameter (n = 7).
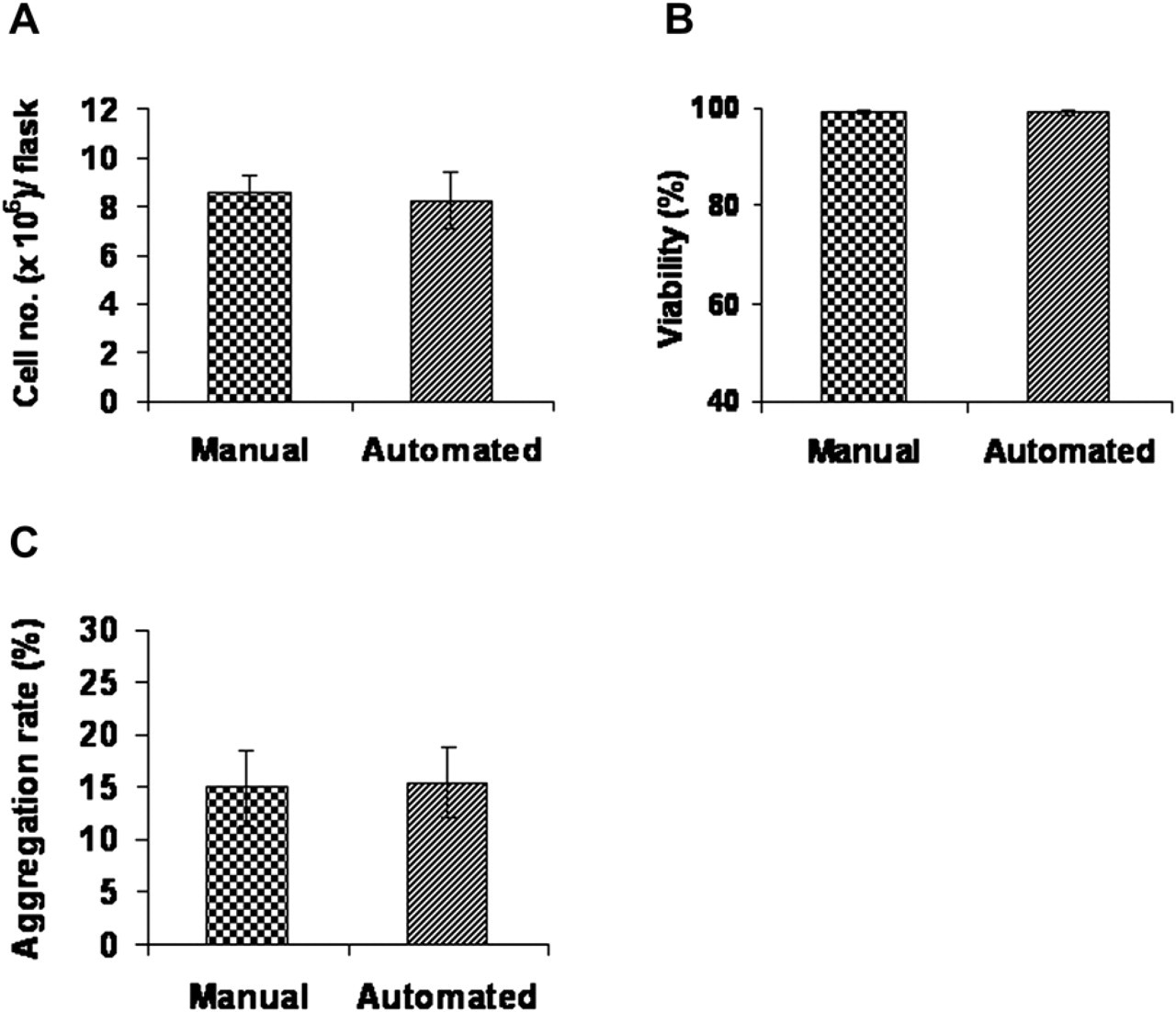
Cells obtained after passages. Isolated disc cells were cultured for 7 days, split and seeded at 2,000 cells/cm2. When cell confluency was estimated to be about 70%, cells were harvested and analyzed (passage 2). Cells from seven flasks of a representative experiment were analyzed. (n = 7). Cell amount is shown in (A), cell viability in (B), and aggregation rate in (C).
Cell Confluence and Cell Number
Confluence measurements have been established using the Cellavista tool after entering measurement parameters that have to be adapted for the respective cell morphology. In this process, cells are detected as objects and the area covered by objects (=cells) can be determined (=confluence).
Cell confluence was correlated to cell number by measuring confluence in RoboFlasks followed by trypsinization and cell counting (n = 5). Comparing cell numbers with the respective confluence measurement indicated a linear correlation if the covered area was between 15% and 70% (Fig. 4). On the basis of this result, total cell numbers can be estimated from the confluence measurement in a noninvasive manner.
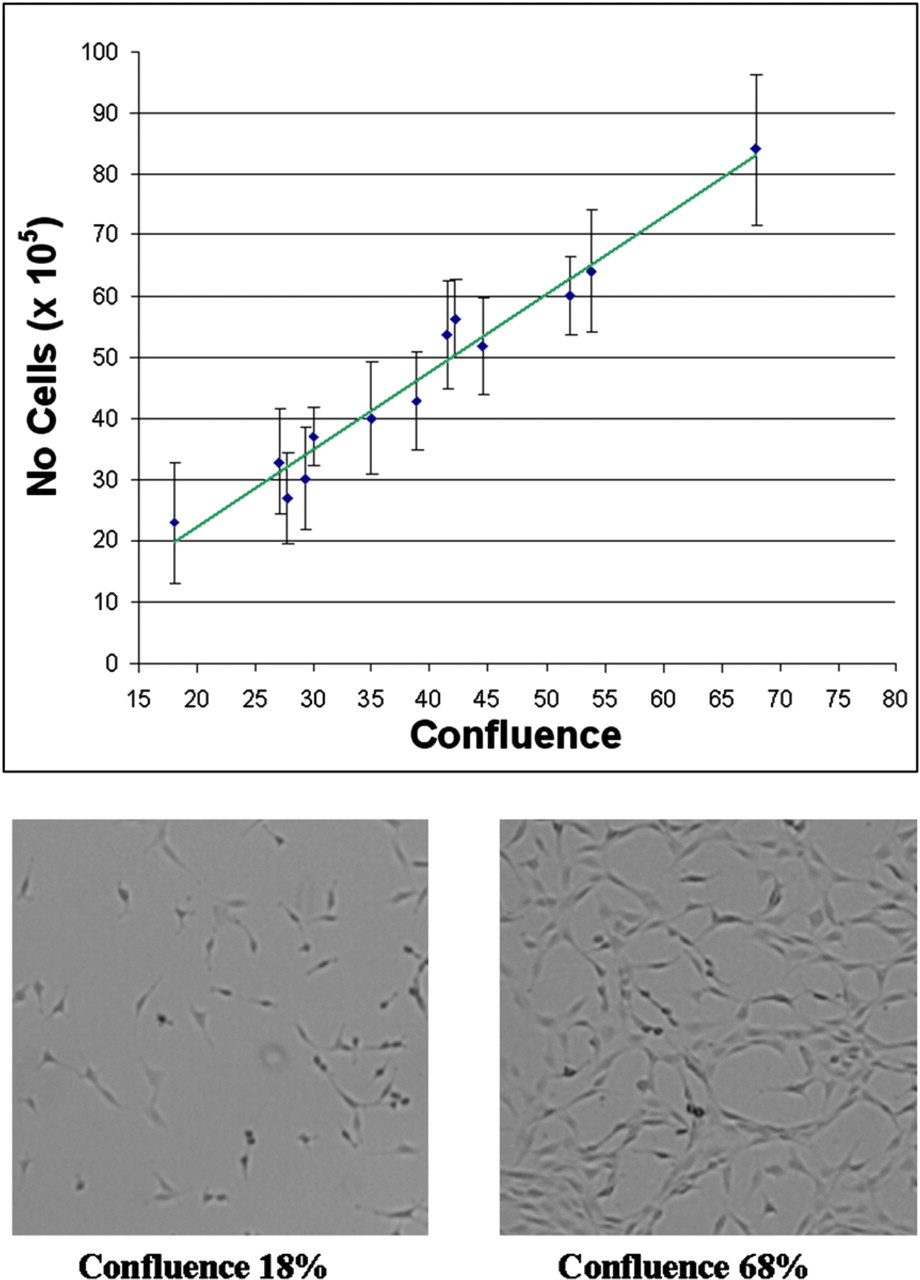
Cell confluence-cell amount correlation. Disc cells were seeded in RoboFlasks in increasing cell densities (1,000–15,000 cells/cm2) and cultured for 3 days. Confluence was measured and cells counted after harvesting. Plotting the cell confluence against the cell amount, a trend correlating the cell confluence with the cell amount was identified. Confluence measurement is the mean ± standard error of the mean from 20 images/RoboFlask analyzed by Cellavista. Representative microscopic images of cell confluence of 18% and 68% photographed by Cellavista are shown.
Growth Rates
In Figure 5, we illustrate the confluence measurement to determine growth rates noninvasively. Cells from the same biopsy were isolated and expanded manually or in the automated system, and confluence was measured continuously during the culture process at days 3, 4, 5, and 6. In both systems, the growth rate has been determined between days 3 and 6, with no difference between the two procedures.
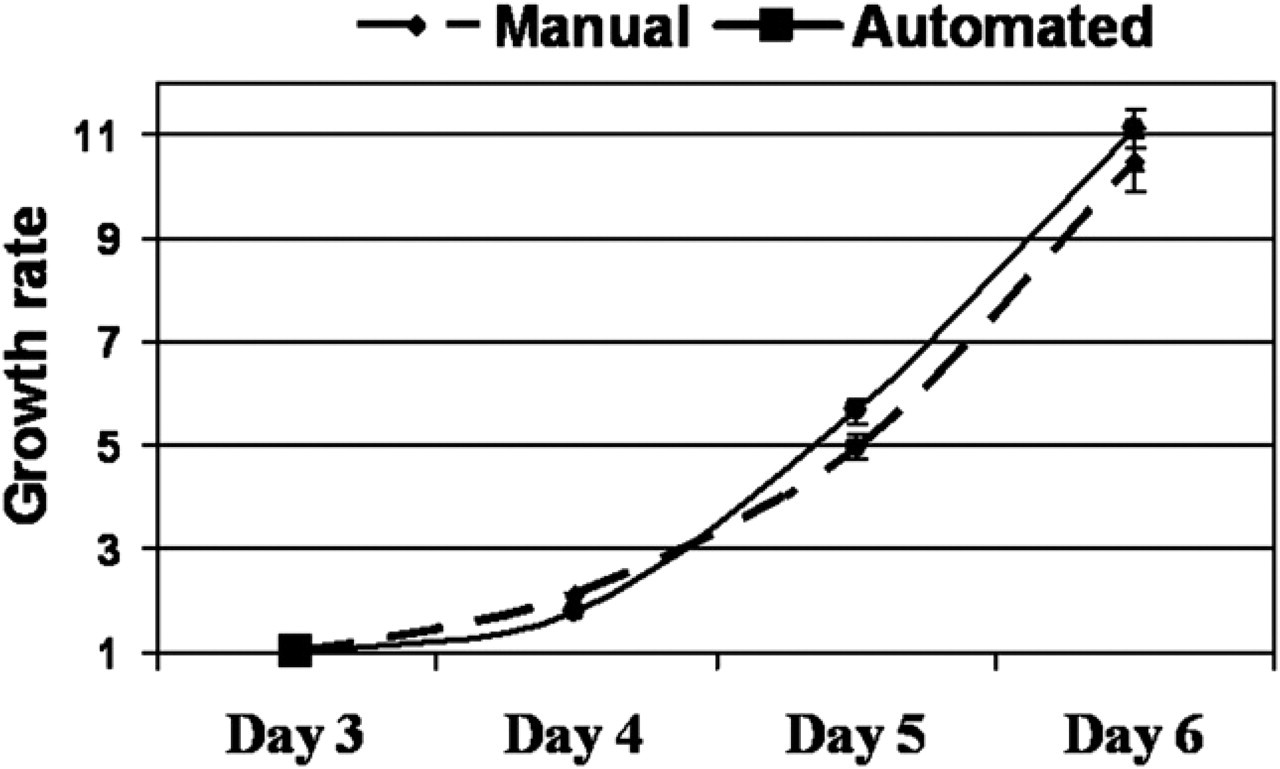
Cell growth rate. Cell growth rate of representative automated or manually isolated cells from the same biopsy is calculated by dividing the confluence measured daily after 3, 4, 5, and 6 culture days by the confluence measured at day 3. Results are expressed as mean ± standard error of the mean (mean confluence is measured by analyzing 20 images/RoboFlask).
As growth rates are not only cell-type specific, but may also be donor dependent, we determined the time needed to reach a certain target confluence (here: 50%) in different donors seeded with the same initial seeding density (2,000 cells/cm2). As shown in Figure 6 for two exemplary donors, cells from different donors can require different time periods to reach the same confluence level, despite the same seeding density and culture conditions (6 vs 7 days to reach 50% confluence). By regular automated confluence measurement, it is possible to harvest the cells at a desired confluence, as shown by Figure 6, where the target confluence was set to 50%.
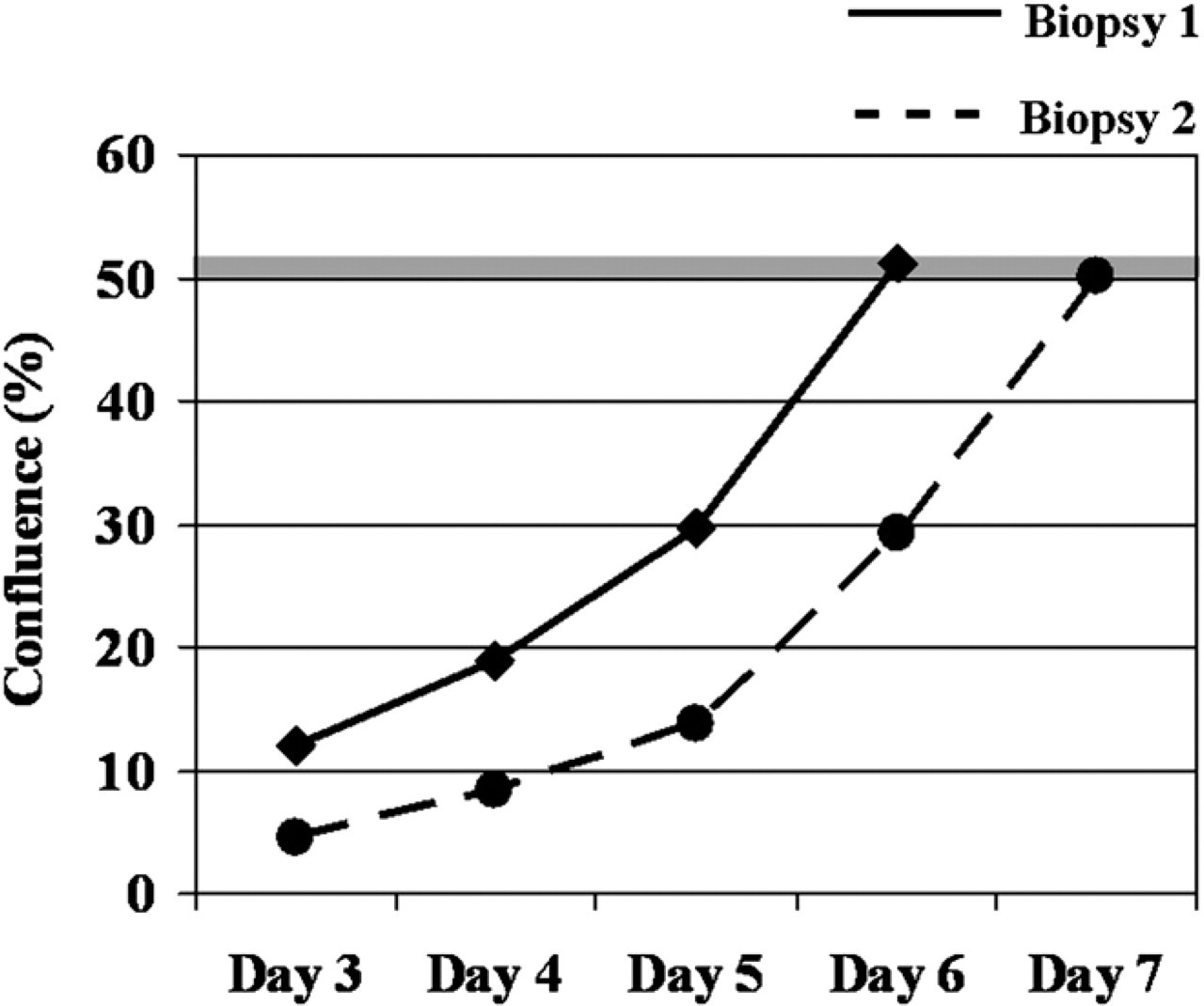
Automated decision on cell splitting. In this example, the cell confluence kinetic of cells isolated from two biopsies is shown. The software of the automated instrument was set to start the harvesting process when the measured cell confluence attains 50%. For biopsy 1, this level occurred at culture day 5, for biopsy 2 at day 6.
Phenotype Characterization
As stability of the cellular phenotype during expansion is important for quality-control purposes, phenotypic characteristics in manually or automatically isolated and expanded cells were compared using immunofluorescence staining for the markers collagen type I and II, versican and total proteoglycan. Our results (Fig. 7A) show that manually and automatically treated cells show a similar phenotype, with expression of all chosen markers. The developed instrument performs the immunostaining of samples in a similar manner to the manual one, as shown in Figure 7B for collagen I and II. Finally, the integrated Cellavista tool allows the automated detection of immunostained samples, as shown in Figure 7C for collagen II, leading to similar signal intensities irrespective whether the cells were manually or automatically isolated and cultured (Fig. 7C).
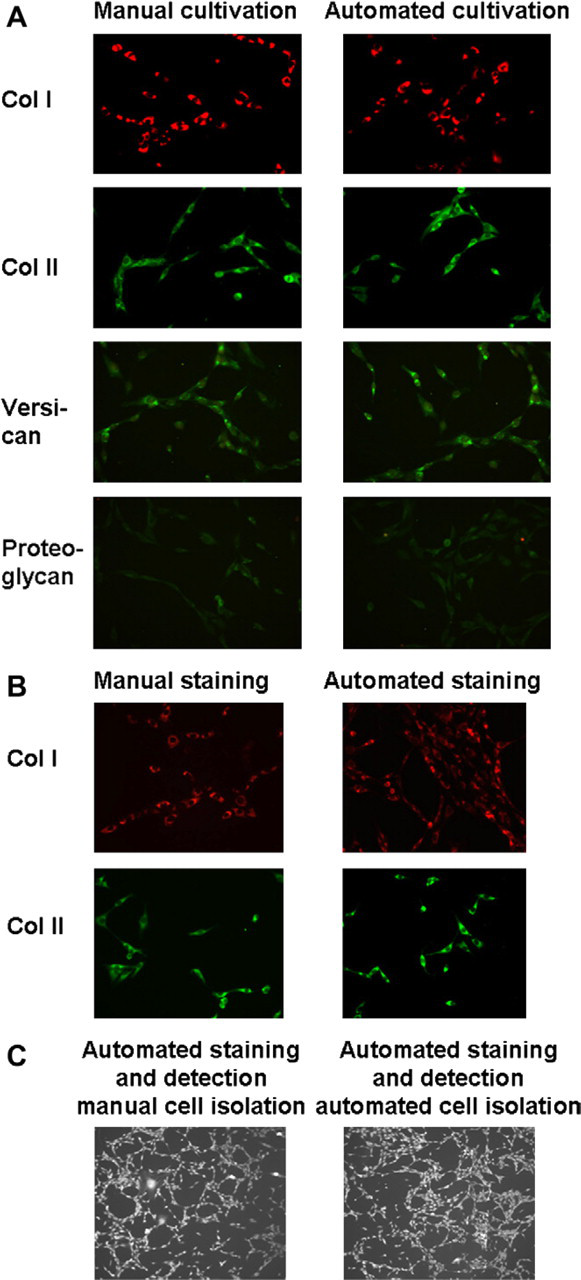
Marker analysis. A. Representative immunofluorescent staining for collagen I, collagen II, versican, and total proteoglycan of cells isolated by manual or automated procedure is shown. Magnification 20×. B. Manual and automated immunofluorescent cell staining for collagens I and II. Magnification 20 ×. C. Automated detection and automated immunofluorescent staining for collagen II of manually and automatically isolated disc cells. Magnification 10 ×.
Discussion
Development of tools for up-scaled culture and expansion of human cells is currently one of the most important challenges in the field of bioengineering, as it is a prerequisite for regenerative medicine to become a clinical reality over a broad spectrum. Only automated systems can provide safe, reproducible, effective, and affordable cell-based products that are able to meet legislative needs such as good manufacturing practice (GMP) requirements. 2
There is already some published work on automated cell-culture platforms, 4 –8 indicating that automation can replace manual procedures for some steps of the cell-culture process. Our results confirm these earlier findings as they demonstrate that the growth and phenotypic characteristics of the automated system were comparable to the manual procedure (Figs. 1–3). However, literature discussing existing automated systems pointed out that “implementation of a centrifuge and automated optical read-out systems for cell density, morphology, and fluorescence” 6 is an important prerequisite for the automated platforms to be developed. Therefore, not only a centrifuge, but also three additional novel features have been successfully implemented into our automated system: (1) a tool to homogenize the biopsy and to isolate the cells automatically, (2) a tool to determine confluence and total cell number automatically and without altering the cells, and (3) a tool to perform marker analysis for quality control. These three inventions resulted in a significant improvement compared with previously presented automation platforms as described below.
Although feeding, passaging, and harvesting of cells are often included in existing platforms, cell isolation from the biopsy is commonly missing in the automated process, although this is particularly important as it reduces the risks that arise from manual handling of a potentially hazardous human biopsy. To the authors' knowledge, this has only so far been achieved in the Celution system (Cytori, San Diego, CA), which specializes in stem cell isolation from adipose tissue. 8 However, for most connective tissues (e.g., cartilage, disc, muscle, ligament), a mincing step is required, which is now available in the system described in this article. We were able to demonstrate that automated isolation using the Dispomix tool results in an equally high cell number with comparable viability and morphology (Fig. 1) while almost completely avoiding any handling of the biopsy by laboratory personnel.
For most clinical applications, a minimal cell number has to be obtained before the patients can be treated (e.g., a certain cell density to be seeded into an implant). So far, to determine an accurate cell number, cells had to be harvested (e.g., via trypsin treatment), thus resulting in increased stress and passage numbers. Using the Cellavista tool in an automated platform not only allows the user to determine how confluent cells are (Figs. 4 and 6), but also to accurately estimate total cell numbers if the cell-type specific correlation between cell numbers per area and confluence has been previously entered into the system. Importantly, no interference with the cells is necessary as the measurement can be done in the Robo-Flask by simply moving it from the incubator into the Cellavista analyzer using the robotic arm. If performed regularly during the culture period, growth rates can be calculated from the saved data (Fig. 5), reflecting the real proliferation of the cells, which is an important quality control aspect (see below). In contrast to other systems, our system does not use standard values for a certain cell type, but calculates each parameter individually for cells from each donor, thus taking into consideration typical donor-donor variations seen for primary cells (Fig. 6).
Safety of a cell-based clinical product can only be achieved if quality control is implemented at least at the end of the process. When using an adequate, cell-specific set of criteria, a decision-making step can be created that helps to provide a clear justification for either usage or rejection of the cells. Similarly, process control can be included during the culture period to achieve consistent quality of the final therapeutic product. We have achieved this via integration of the cell analysis platform Cellavista, which allows image-based, noninvasive evaluation of the cells, either using brightfield or fluorescent illumination.
Brightfield analysis enables calculation of growth rates (Fig. 5) (as described above), which can be used for quality control as (cell-specific) untypically low growth rates indicate alterations in the cellular metabolism. An automated warning can be issued by the system, so that the user can decide whether to continue or to stop the culture.
Fluorescence analysis can be used to detect cell-specific markers that either indicate normal phenotypic characteristics or, incontrast, indicate unwanted dedifferentiation of fully differentiated cells or unwanted differentiation of progenitor cells. In our experiments, we used specific antibodies for typical disc cell markers such as total proteoglycans, collagen, or versican and visualized the expression using fluorescence-labeled secondary antibodies. As no specific marker is present for nucleus pulposus or annulus fibrosus cells, we used a panel of markers that covers the extracellular protein profile of these cells. We were able to demonstrate that the chosen markers can be detected to the same degree in cells from the automated system and the manual procedure (Fig. 7A). As different color labeling of the secondary antibody is possible, various important markers (specifically chosen for each cell type) can be detected simultaneously if the software is adjusted accordingly and if the staining procedure is established. As for the growth rate, an automated warning can be issued if the marker profile does not meet the set requirements (e.g., occurrence of an unwanted marker).
In our experiments, we performed medium-term culture for 14 days, which is equivalent to two passages. During this time, the system was free of any contamination and no differences in cell number, viability, or morphology could be observed between the automated and manual procedure. No further passages have been investigated as they are usually avoided for disc cells because these are very susceptible to dedifferentiation during 2D expansion. 15 Although our results indicate that longer culture periods should be possible (if necessary for other cell types), this remains to be verified in the future.
In summary, this novel platform enables isolation and culture of human primary (disc)cells, with similaryields, viability, and phenotype compared with the manual procedure. Important steps such as user-safe handling of the biopsy and quality control methods have been implemented so that the platform can be used to replace the conventional manual methods. Importantly, instead of an established cell line, primary cells have beenused as they “are more sensitive to process variability (…) and present a greater technical challenge (…) in regenerative medicine.” 7 Human disc cells were used as a model cell type, but the system could be easily adapted to other primary cells with only minor modifications, for example, entering the cell-dependent specifications for trypsin treatment.
Acknowledgments
This project was funded by CTI (Commission for Technology and Innovation of the Swiss Confederation) and Tecan Schweiz AG.
We thankfully acknowledge the assistance of Sybille Grad, AO Research Institute, AO Foundation, Davos, Switzerland, for helping us in the choose of the phenotypic markers and related antibodies.
Competing Interests Statement: The authors certify that they have no relevant financial interests in this article and that any/all financial and material support for this research and work are clearly identified in the article.