
Abstract
Aqueous solubility is an important physicochemical parameter for any potential drug candidate, and high-throughput kinetic assays are frequently used in drug discovery to give an estimate of a compound's aqueous solubility. However, the aqueous solubility data from an equilibrium (thermodynamic) shake-flask technique is considered more relevant, but is slower and more labor intensive to generate. A highly automated aqueous equilibrium solubility shake-flask technique is described and validated on a set of 15 marketed drugs, whose aqueous solubilities cover four orders of magnitude. The assay uses a Tecan Freedom Evo 200 liquid handling robot (Tecan Group Ltd., Männerdorf, Switzerland) with integrated appliances for the transportation, decapping and recapping, and centrifugation of sample tubes. These bespoke automation solutions help overcome the labor intensive steps associated with performing conventional, gold standard, aqueous equilibrium solubility shake-flask measurements, enabling the assay to be used as a primary-wave drug discovery screen.
Introduction
Only the compound in a solution can be absorbed into the systemic circulation; hence, in vivo efficacy for oral or inhaled (dry powder) therapies is dependent on a compound's dissolution into aqueous biological media.1,2 Dissolution into gastrointestinal or lung fluid will be dependent on many factors, including physicochemical, physiological, and formulation parameters. 3 Furthermore, most in vitro and animal in vivo experiments require a compound to have a certain degree of solubility in an aqueous media. An early indication of a drug's ability to dissolve in aqueous media is highly desirable from a drug discovery perspective.
Typically, equilibrium (thermodynamic) or kinetic aqueous solubilities are determined. The aqueous equilibrium solubility is the solubility that is observed once equilibrium has been achieved between the solution and solid material. Equilibrium may be reached quickly or may take several days to achieve. Kinetic aqueous solubility is the solubility of the compound at a particular time within the aqueous medium when the system may not be at its equilibrium position. 4
Kerns et al. 5 recently reviewed the current methodology used within the drug discovery context and highlighted that aqueous solubility assays can be broadly split into either kinetic or equilibrium assay. One key difference between these two techniques is the starting form of the compound. Some kinetic assays involve addition of the compound to an aqueous medium after predissolution in an organic cosolvent, for example, dimethylsulfoxide (DMSO), thereby removing the effect of crystal form on a compound's ability to dissolve. In contrast, equilibrium assays tend to differ, with the aqueous media being added to the solid form of the compound. Thus, the solubility measurement will not be altered by the presence of a cosolvent. Equilibrium shake-flask aqueous solubility assays are used in drug development and are generally considered to be the gold standard technique. 5 It involves the solid being placed in a vial to which the aqueous media is added and the vial then sealed and shaken. After a sufficient period of time, when the system has likely reached its equilibrium position, any undis-solved compound is removed, typically using filtration, and the concentration of the compound in the filtrate is determined. The use of filters can perturb the equilibrium, as dissolved compound can be adsorbed on to the filter during filtration, lowering its concentration in the filtrate. 6 This can be a concern for compounds with borderline aqueous solubility (e.g., <10 μM). An alternative separation is to use a double-centrifugation technique. This involves the centrifugation of the suspension to pellet out any undissolved compound followed by the transfer of the supernatant to another vial and its subsequent repeat centrifugation. This second centrifugation is necessary to ensure that any remaining suspended particulates are pelleted out, allowing an aliquot of the supernatant to be removed and analyzed.
An estimation of a compound's aqueous solubility in the very first stages of the drug discovery screening cascade is common practice. It is important to realize that the challenges facing an aqueous solubility assay in these early stages of drug discovery are different from those encountered during the drug development process. 7 Drug discovery aqueous solubility assays need to be able to handle large numbers (∼96) of diverse compounds on a weekly basis. The amount of compound available at this stage may be small (1-2 mg), and the solid-state properties of each batch of compound will typically be uncharacterized. The main aim of this measurement is to provide an estimate of the aqueous solubility of a compound, so that an informed decision can be made as to whether the current batch of the compound has the desired properties that will enable it to progress further. In addition, the estimate of a compound's aqueous solubility needs to be consistent with the more detailed aqueous solubility measurements that will eventually be performed if the compound is considered for testing in safety and/or in vivo disease models.
Herein, we present the development of an automated aqueous equilibrium solubility shake-flask assay. It uses a 66-h equilibration time between a solid sample and aqueous buffer, a double-centrifugation separation technique, and easily supports up to 96 compounds. The assay makes use of a Tecan Freedom Evo 200 liquid handling robot with integrated appliances for the transportation, decapping and recapping, and centrifugation of sample tubes. These bespoke automation solutions help overcome the labor intensive steps that would normally prohibit the use of a conventional, gold standard, aqueous equilibrium solubility shake-flask technique as a primary-wave drug discovery screen, thus enabling aqueous equilibrium solubility measurements to be performed on all newly synthesized compounds with a turnaround time of six calendar days from receipt of samples.
Rationale for the Development of the Automated Assay
Before the development of the automated aqueous equilibrium solubility assay, a low-throughput (24 compounds per assay) shake-flask technique for assessing the aqueous equilibrium solubility of drug discovery compounds had been performed in our laboratory. This involved adding pH 7.4, 0.1 M phosphate buffer to a solid sample of compound, shaking for a minimum of 18 h at 20 °C, and then analyzing the compound concentration in the supernatant after a double-centrifugation step.
The decision to determine aqueous solubilities in a pH 7.4, 0.1 M phosphate buffer was based on the fact that most subsequent in vitro assays (e.g., potency, plasma protein binding, and others) used similar media and require compounds to have a minimum solubility. Knowledge that a compound has limited solubility in a relevant buffer is useful in deciding how to work with it in subsequent in vitro assays. The high ionic strength of the pH 7.4, 0.1 M phosphate buffer provides good buffering capacity for highly soluble compounds that are readily ionized in solution. The use of such a buffer can result in basic compounds, when present at high concentrations, precipitating out as a phosphate salt. Consequently, the applicability of the Henderson—Hasselbalch equation to the estimation of the solubility—pH profile of such compounds using such solubility data should be done with caution. 8 However, assessing the likelihood of solubility issues in subsequent in vitro assays is highly relevant information to gain from a primary-wave drug discovery assay, and solubility—pH profiles can be determined at a later stage in more appropriate buffer systems.
The low-throughput shake-flask technique lacked the capacity to support all newly synthesized compounds. A 96-well filter plate—based technique was also developed and run for 6 years typically on a weekly basis. This technique involved adding an aliquot of compound predissolved in DMSO to pH 7.4,0.1 M phosphate buffer followed by shaking for a minimum of 18 h at 20 °C before filtering and analyzing the compound concentration in the filtrate. The upper limit for this filter plate assay was approximately 100 μM (i.e., 0.05 mgmL−1), and during this time, the low-throughput shake-flask technique was run as an additional later-stage (secondary or tertiary wave) assay for determining the aqueous solubility of compounds that had progressed further through a drug discovery screening cascade. For comparison, the upper limit of the low-throughput shake-flask technique was approximately 2200 μM (about 1.1 mg mL−1 for typical molecular weight compounds). With time, it became apparent that the two assays were generally providing similar information, but there was a significant degree of variability between the two measurements. This is shown in Figure 1, which shows the plots for the pH 7.4, 0.1 M phosphate buffer solubility of compounds measured using the low-throughput shake-flask technique compared with the filter plate technique that uses either an oleophobic polykryptonite (PKP) or glass microfibre (GF/B) filter. Although the data displayed in these plots show only moderate correlations, the relationships still show high statistical significance, which is indicative that the two different techniques are providing some similar information. As the low-throughput shake-flask technique was more similar to aqueous equilibrium solubility techniques that are performed in drug development, it was considered to be a better estimate of a compound's aqueous equilibrium solubility. The deviations from these values when measured in the filter plate assay could be rationalized in a number of ways. These include the nonspecific binding to the filter
6
or formation of supersaturated solutions with slow precipitation kinetics or cosolvent effects.
9
It was felt that the aqueous solubility data generated using the filter plate technique was too different from that using the low-throughput shake-flask technique for our purposes. Using the filter plate technique to identify poorly aqueous solubility compound carried an unacceptably high risk of progressing false-positive and missing true-positive compounds. Furthermore, it was deemed inappropriate to use the filter plate aqueous solubility data to build a quantitative structure-property relationship (QSPR) model for aqueous equilibrium solubility with sufficient predictive power for compound design.
10
The data generated using the filter plate technique was able to provide an approximate estimate of a compound's aqueous solubility and was helpful in troubleshooting issues seen in in vitro assays where a DMSO liquid stock of the compound is used. However, the decision was taken to abandon the filter plate aqueous solubility technique and just measure aqueous solubility using a shake-flask technique, which had the additional benefit of a higher upper limit.
Aqueous solubility (pH 7.4) data measured using a low-throughput shake-flask technique compared with those using the abandoned high-throughput 96-well (A) PKP (n = 168) or (B) GF/B (n = 287) filter plate technique.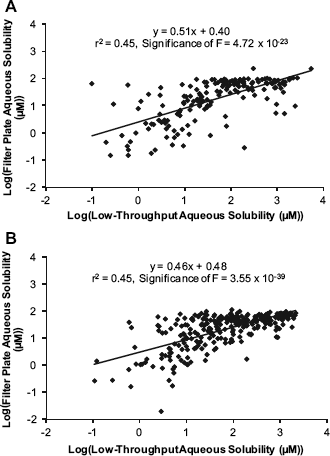
The existing low-throughput shake-flask technique was very labor intensive and lacked the necessary capacity to be realistically used as a primary wave screen within a drug discovery screening cascade. The essential steps involved shaking a solid sample of a compound in an aqueous buffer within a sealed receptacle, after which the aqueous suspensions were centrifuged twice, and an aliquot of the second supernatant was removed and analyzed by HPLC using ultraviolet (UV) quantification of the chromatographic peak that had been identified using MS. To address the low throughput, all liquid handling steps were transferred to a liquid handling robot fitted with an eight-disposable-tip configuration. Shaking could be carried out using an integrated T-shake unit but was predominantly carried out using a standalone IKA KS 260 orbital shaker (IKA Werke GmbH & Co. KG, Staufen, Germany) because of its increased capacity. The centrifugation process was transferred to an integrated Hettich Rotanta 46 RSC centrifuge (Andreas Hettich GmbH & Co. KG, Tuttlingen, Germany). Two integrated robotic manipulator arms (ROMAs) moved samples around the liquid handling robot. One of these ROMAs had extended manoeuvrability in the z-axis and was primarily used to transfer samples to and from the integrated centrifuge situated under the main work surface. The rate-limiting step in the automation was the decapping and recapping of receptacles before and after a liquid handling step. Tecan Group Ltd. was commissioned to integrate an Abgene ALTO-8 de-capper unit (Abgene Ltd., Epsom, Surrey, UK) onto the liquid handling robot. This decapper unit was able to programmatically decap a column of eight 1-mL, polypropylene, twist-lock tubes in 96-well format, and then allow a liquid handling arm (LIHA) to dispense or aspirate aqueous samples from or into the decapped tubes, after which the tubes could be recapped. The Twist-Lock tubes consisted of a polypropylene tube with a chemically resistant thermoplastic elastomer cap that provides a seal to sample loss.
A concern with the development of this higher-throughput shake-flask assay was the increased need for the weighing out of solid samples. In our experience, there was a clear benefit in the quality of the aqueous solubility data from using solid starting material compared with starting from the more easily dispensed DMSO liquid stock. We, therefore, justified the extra effort that was involved in the dispensing of solid starting material. We acknowledge that, for other groups, a similar cost—benefit analysis may prohibit such additional work. A possible compromise may be the use of solid material derived from dried DMSO liquid stocks, and such work is beyond the scope of this manuscript.
Experimental
Instrumentation and Analysis
All pre— and post—data handling procedures were carried out using custom built Microsoft Excel workbook templates and add-ins. Automated liquid sample handling was performed using the liquid handling robot fitted with an eight-tip configuration that used standard disposable tips. The liquid handling robot was fitted with two ROMAs. Both ROM As were used to move sample plates around the robot's work surface, and one was able to access an integrated temperature-controlled (20 °C) centrifuge positioned below the robot's work surface (i.e., zROMA). Additional integrated appliances on the liquid handling robot included a T-vac unit, for filtering 96-well filter plates; a T-shake unit, for shaking 96-well format plates; and a screw-top decapper unit. Figure 2 shows a photograph of the liquid handling robot along with the integrated appliances described earlier. The liquid handling robot and all its integrated appliances were controlled using Tecan Gemini software (Tecan Group Ltd.). Any manual liquid handling was performed using Eppendorf electronic pipettes fitted with Eppendorf disposable tips (Eppendorf UK Ltd., Cambridge, Cambridgeshire, UK). Centrifugations for the low-throughput shake-flask assay were carried out using a temperature-controlled (20 °C) Heraeus Biofuge Fresco (Thermo Fisher Scientific, Loughborough, Leicestershire, UK). For the high-throughput shake-flask technique, the T-shake unit could be used, although the shaking of samples for all three techniques was predominantly carried out using an orbital shaker set at 450 rpm. This orbital shaker had an increased capacity and allowed for the plates containing the samples to be turned on their side for shaking. All HPLC analyses were carried out using a Waters 2690 or Waters 2695 separation module (Waters Ltd., Elstree, Hertfordshire, UK) and Waters 2777 autosampler (Waters Ltd.). The autosampler sample holder was fitted with a Peltier cooler set to 20 °C. A Waters 996 diode array detector (Waters, Ltd.) was used for UV detection, and MS detection was carried out using either a Micromass ZQ (Waters Ltd.) or a Micromass Quattro Micro (Waters Ltd.) in scan mode. Both UV and MS data were simultaneously collected for each HPLC sample. Before a UV chromatographic peak was quantified, the corresponding MS trace was manually inspected to confirm the presence of a corresponding MS peak for the compound. If the MS trace failed to show the correct MS peak, no aqueous solubility value was reported. Waters Quanlynx software (Waters Ltd.) was used to process all the HPLC/UV chromatograms. Typically, UV quantification was performed at 260 nm; when the response was poor or saturated the UV detector (i.e., > 1 absorbance unit), an alternative UV wavelength was used (if necessary, further dilutions were made and analyzed). Waters Symmetry C8 3.5-mm, 4.6 × 50-mm columns (Waters Ltd.) were used for HPLC chromatography along with a gradient of 1% acetonitrile: 99% 0.1% aqueous formic acid to 99% acetonitrile: 1 % 0.1% aqueous formic acid at a flow rate of 2 mL min−1 over 3.5 min. All pH measurements were conducted using a Fisher Scientific AR20 pH/conductivity meter (Thermo Fisher Scientific). All experiments were carried out in constant temperature laboratory held at 20 °C. A MilliQ water purification system (Millipore Corporation, Billerica, MA) was used to supply all water for experiments.
Photograph of the liquid handling robot setup.
Materials
All compound samples were weighed out into vials by hand. Abgene twist-lock 1-mL capped tubes in 96-well racks, Whatman Unifilter (glass-filled polypropylene, 2 mL, 96 well) PKP and GF/B filter plates were obtained, along with all other consumables, from Thermo Fisher Scientific. The pH 7.4, 0.1 M phosphate buffer was prepared by dissolving sodium dihydrogen orthophosphate (3.12 g) and di-sodium hydrogen orthophosphate dihydrate (14.24 g) in water (1 L) and, if necessary, adjusted with concentrated hydrochloric acid or sodium hydroxide.
Comparative Low-Throughput Shake-Flask Technique—Low Automation
All liquid handling was carried out using a liquid handling robot. Centrifugation and shaking steps were performed on appliances not integrated onto the liquid handling robot. Decapping of tubes was performed manually.
Up to 24 compounds (1.0 mg) were placed into separate 2-mL glass screw-top vials, to which was added pH 7.4, 0.1 M phosphate buffer (1000 μL). The vials were shaken for a minimum of 18 h. After shaking, the saturated solutions were transferred to 2-mL centrifuge tubes and centrifuged at 13,000 rpm for about 15 min. The supernatants were then removed, placed into new centrifuge tubes, and centrifuged again at 13,000 rpm for 15 min. Aliquots of the supernatants from the second centrifugation were transferred into 1-mL glass vials within a 96-well analysis plate. A 10-fold dilution of each supernatant, using pH 7.4, 0.1 M phosphate buffer, was prepared in 1-mL glass vials within the 96-well analysis plate. Standard stock solutions of the compounds were prepared by adding DMSO (800 μL) to compound (1.0 mg) and sonicating (15 min). If any particular compound had not dissolved after this time, then it was further sonicated (15 min). If it was still not fully dissolved, then an aqueous solubility measurement was not reported for this compound. An aliquot (8 μL) of each DMSO stock solution was added to a 1-mL glass vial within the 96-well analysis plate, to which was added DMSO (792 μL). The solutions in the 96-well plate were then analyzed using HPLC/UV quantification with MS confirmation of each UV peak. The aqueous solubility was calculated from the observed peak area in the HPLC/UV chromatograms along with corrections for any dilutions of the sample and standard and differences in injection volumes using Eq. (1).

Where PAspl and PAstd are the integrated HPLC UV peak areas for the sample and standard, respectively. DFspl is the dilution factor for the sample. IVspl and IVstd are the HPLC injection volumes μL) for the sample and standard, respectively. Zstd is the concentration μM) of the standard solution. If the aqueous solubility is greater than or equal to 110% (allowing for experimental error in the UV response) of the mass (mg) of compound added to the pH 7.4, 0.1 M phosphate buffer divided by the buffer's volume (mL), then the aqueous solubility was reported as greater than this theoretical maximum. Depending on the compound, this was approximately greater than 2200 μM (i.e., > l.l mg mL−1). The lower limit for this aqueous solubility assay is approximately 0.2 μM depending on the UV chromophore of the particular compound.
High-Throughput Shake-Flask Technique—High Automation
The main differences between this technique and the comparative low-throughput shake-flask technique are described as follows. They include the use of the liquid handling robot's integrated decapper and centrifuge combined with ROMAs to move plates between these appliances.
Samples of compounds (1.0 mg) were supplied in twist-lock 1-mL capped tubes within a 96-well plate format. A 96-well plate contained a maximum of 48 compounds dispensed into the first, third, fifth, seventh, ninth, and eleventh columns. For 96 compounds, two plates were used. pH 7.4, 0.1 M phosphate buffer (750 μL) was added to all the wells in the first, third, fifth, seventh, ninth, and eleventh columns of the 96-well plate. This process was fully automated and involved the decapper removing the caps from each column of tubes, the LIHA of the liquid handling robot dispensing the buffer into the uncapped tubes, and the decapper recapping the filled tubes. The process was repeated for each odd column. The plate was then shaken for 66 h. After shaking, the plate was centrifuged at 1000 rpm for 1 h. The supernatant (500 μL) was then removed and placed in the next column of the empty column of tubes. Supernatants from the first column were transferred to the second column; supernatants from the third column were transferred into the fourth, and so on. This process was fully automated and involved the decapper removing the caps from each column of tubes; the LIHA of the liquid handling robot would aspirate from the relevant column and hold the supernatant while the decapper went through the process of recapping the aspirated tubes and then decapping the next column of empty tubes. The supernatants were then dispensed into these empty tubes, and then, the decapper would recap the tubes. The liquid class controlling the aspiration of the supernatant was such that the aspiration position of the disposable tips was maintained at a few millimeters below the surface of the supernatant. This minimized the chances of aspirating solids, which had the tendency to float on the surface of the supernatant, for compounds that formed poorly wetting material. The plate was then centrifuged at 1000 rpm for 1 h. Aliquots of the supernatants (200 μL) from the second centrifugation were transferred into 1-mL glass vials within a 96-well analysis plate. A 10-fold dilution of each supernatant was also prepared, in 1-mL glass vials within the 96-well analysis plate, by diluting an aliquot of the supernatant (25 μL) with pH 7.4, 0.1 M phosphate buffer (225 μL). Standard stock solutions of the compounds were prepared in a 96-well plate by diluting a 20 mM DMSO stock of compound (5 μL) with DMSO (795 μL). The concentration of the standard solution was 125 μM. Analysis of samples and the aqueous solubility determinations were performed in an identical way to that described in the comparative low-throughput shake-flask technique.
Comparative High-Throughput Filter Plate Technique—Low Automation
This technique is similar to other published methods.5,11 Filtration was performed using an integrated T-vac unit on the liquid handling robot, whereas the decapping of tubes was performed manually.
Five milligrams per milliliters of DMSO stock solutions (200 μL) were prepared. An aliquot (8 μL) of each DMSO stock solution was transferred to a 96-well PKP filter plate or a 96-well plate of 1-mL glass vials if a GF/B filter plate was being used. An aliquot of pH 7.4, 0.1 M phosphate buffer (792 μL) was added to all wells in the PKP filter plate or the glass vial plate. The filled 96-well plate was then sealed and shaken for a minimum of 18 h. (Note, unlike the PKP filter plate, the GF/B filter plate that we used had a tendency to leak from the filter over the shaking period, which is the reason for shaking in glass vials.) If a GF/B filter plate was being used, then the buffer solutions were transferred from their positions in the 96-well glass vial plate to corresponding wells in a GF/B filter plate positioned on the T-vac unit above a 96-well plate containing empty 1-mL glass vials. If a PKP filter plate was being used, then the filter plate was positioned on the T-vac unit above a 96-well plate containing empty 1-mL glass vials. A vacuum was applied (5 min), and the samples were collected in the 96-well acceptor plate. A corresponding standards plate was prepared by adding DMSO (792 μL) to 96-well plate containing an aliquot of the DMSO stock solutions (8 μL) in 1-mL glass vials.
Analysis of samples was performed in an identical way to that described in the comparative low-throughput shake-flask technique. The aqueous solubility was calculated using Eq. (2).
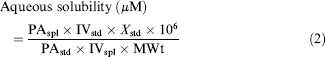
Xstd is the concentration of the standard solution (0.05 mg mL−1). MWt is the compound's molecular weight. This method has a top limit of approximately 100 μM (exactly 0.05 mg mL−1), corresponding to complete dissolution of the sample, and a lower limit of about 0.2 μM depending on the chromophore of the particular compound.
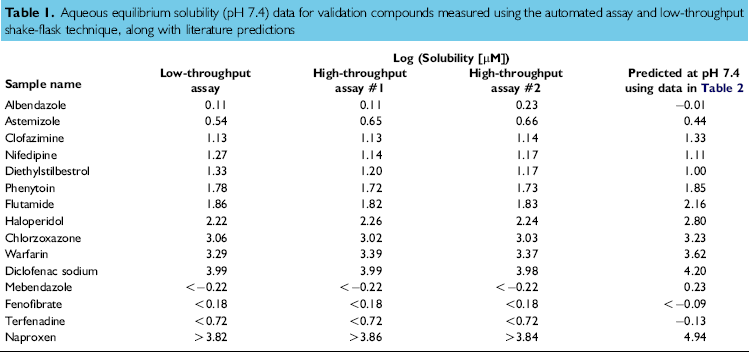
Validation of the Automated Assay
A validation set of 15 compounds, with aqueous solubilities ranging over four orders of magnitude, was chosen. The aqueous equilibrium solubilities were measured using the automated assay in duplicate, in separate assays, to assess the precision of the technique. For comparison, the validation set also had aqueous equilibrium solubility measured in our low-throughput shake-flask assay. All measurements are listed in Table 1. The aqueous equilibrium solubility measurements of mebendazole, fenofibrate, terfenadine, and naproxen were reported as out-of-range values and, subsequently, have been left out of any statistical comparisons. For the purposes of this validation, the shaking time for both techniques in this comparative study was kept to 18 h. In searching the literature, it was not possible to find comparative aqueous equilibrium solubility data for all the validation set performed in a pH 7.4 buffer. Instead, predictions of the aqueous solubility at pH 7.4 were made through correction of literature estimates of the intrinsic solubility of these compounds with the use of Eq. (3).
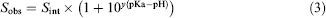
Aqueous equilibrium solubility (pH 7.4) data for validation compounds measured using the automated assay and low-throughput shake-flask technique, along with literature predictions
Equation (3) summarizes the Henderson—Hasselbalch equations for monoacid and monobasic compounds. 12 Sobs is the observed solubility at a particular pH (the sum of the concentration of the ionized and unionized forms of the compound that are in solution). Sint is the intrinsic solubility (the concentration of the unionized form that is in solution). For monoacidic compounds, y = —1, and for monobasic compounds, y= 1. These predictions are listed in Table 1, and the literature intrinsic solubility and ionization data used in these predictions are listed in Table 2.
Literature intrinsic solubility and ionization data used to predict the aqueous solubility of the compounds in Table 1
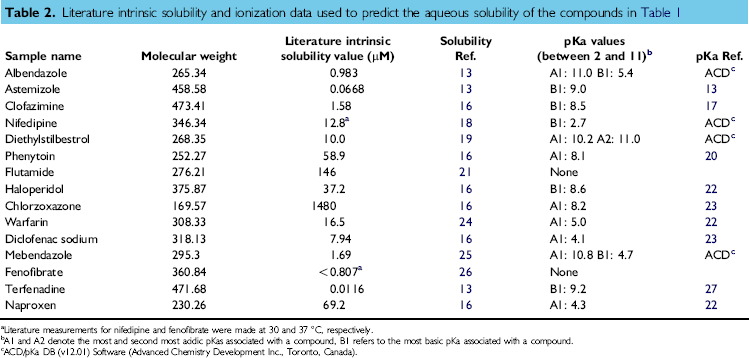
Literature measurements for nifedipine and fenofibrate were made at 30 and 37 °C, respectively.
A1 and A2 denote the most and second most acidic pKas associated with a compound, Bl refers to the most basic pKa associated with a compound.
ACD/pKa DB (vI 2.01) Software (Advanced Chemistry Development Inc., Toronto, Canada).
The correlations shown in Figure 3A and B demonstrate excellent agreement between the logarithms of the aqueous equilibrium solubility values determined using the low-throughput shake-flask technique and those determined from automated assay. The root mean square error for the data shown in Figure 3A is 0.083, and for the data shown in Figure 3B, it is 0.092. Furthermore, there is very good agreement between the aqueous equilibrium solubility data generated by the automated assay and the predictions based on literature intrinsic solubility data. Most of the intrinsic solubility data are measured at room temperature, except for nifedipine and fenofibrate. Nifedipine's intrinsic solubility is 12.8 μM, as measured at 30 °C, and at room temperature, this value would be expected to be smaller. Considering the predicted aqueous solubility to be almost identical to that measured using the automated assay, a two- to threefold reduction in nifedipine's intrinsic solubility in going from 30 °C to room temperature would still be acceptable. Fenofibrate's intrinsic solubility is reported as less than 0.806 μM, as measured at 37 °C; again, at room temperature, this value would be expected to be smaller. Using the automated assay, it was not possible to determine an absolute aqueous equilibrium solubility value because of the lack of UV sensitivity. Hence, a “less than” aqueous equilibrium solubility figure was quoted, which is consistent with the aqueous solubility prediction for fenofibrate at room temperature.
Comparison of the aqueous equilibrium solubility (pH 7.4) measurements made using the low-throughput shake-flask technique compared to those made using the automated assay in two separate attempts (A and B).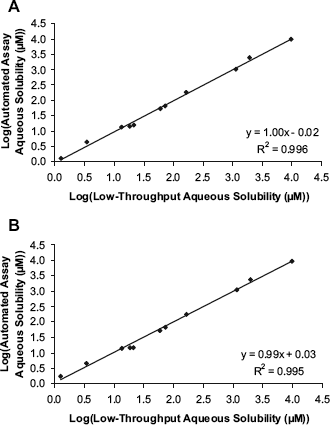
To test the assay-to-assay precision of the automated assay, the aqueous equilibrium solubility of the same batch of glyburide was run in 30 separate assays. These experiments led to a mean aqueous solubility of 9.25 μM and standard deviation of 1.95 μM. Using a literature intrinsic solubility of 0.0891 μM and an acidic pKa of 5.3 in Eq. (3), a consistent value of 11.3 μM for the pH 7.4 aqueous solubility of glyburide is predicted.13,14 Furthermore, a consistent value of 16.6 μM (8.22 μg mL−1) for the aqueous solubility of glyburide, measured at 25 °C, has been reported by Seedher and Kanojia.
15
The spread in the 30 repeat measurements for glyburide can be seen in Figure 4, which also indicates the mean and 95% confidence intervals of the mean (8.52 and 9.98 μM).
Plot of the spread in the 30 repeat aqueous solubility (pH 7.4) measurements for glyburide as measured using the automated assay.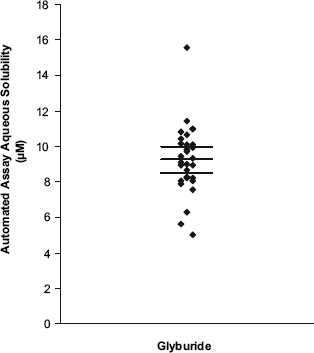
It is important to understand the precision of the automated assay for compounds with borderline aqueous solubility (i.e., < 10 μM), because a decision to progress these compounds may need to be made on a single measurement. Glyburide is a good test compound, and the aqueous equilibrium solubility data show an approximate threefold spread (i.e., ∼5—15 μM) for the repeat measurements (although based on the standard deviation, 95% of repeat measurements are expected to fall within a 2.5-fold spread around the mean). This spread in the repeat data is consistent with the expected quality for an early drug discovery screen. It is assumed that the spread in repeat measurements for more soluble compounds is homoscedastic. In other words, 95% of repeat aqueous solubility measurements for any compound would fall within a 2.5-fold spread around the mean. Although an exhaustive analysis has not been performed, Figure 5 displays a plot of a set of repeat aqueous equilibrium solubility measurements generated using the automated assay on the same batches of compounds but run on different days. A paired t-test indicates that there is no significant difference between these repeat measurements when testing at the 95% significance level. This is further evidence that the automated assay can repeatedly generate reliable aqueous solubility data for diverse compounds.
Repeat aqueous solubility (pH 7.4) data measured on the same sample of a compound, but on different days, using the automated assay.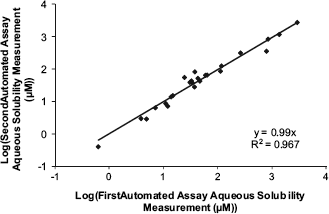
The automated assay uses a double-centrifugation step. In theory, the aqueous solubility measurements will be independent of the number of centrifugation steps. The first centrifugation step will be sufficient in most cases, and the second centrifugation step is, therefore, a precautionary step to mitigate against the chance transfers of undissolved material when aspirating supernatant. We have not conducted any systematic investigation into this, but experience would indicate, there is value in performing a double-centrifugation step for poorly wetting material solids that have the tendency to float on the surface of the supernatant. For these types of solids, the possibility of chance transfers of undissolved material is higher after the first centrifugation step.
Conclusions
Within a typical design-make-test drug discovery process, there are two important reasons for high-quality aqueous solubility measurements. First, drug discovery projects will have clearly defined progression criteria for measured aqueous solubility, and it is indefensible for that compound to have its progression hindered, or stopped, because of the lack of a reliable measurement. Second, these measurements can be used to train QSPR models that can be used to generate predictions and prioritize future targets, and the quality of predictions will decrease as the level of error in the aqueous solubility data increases.
Our highly automated shake-flask technique favors the determination of the aqueous equilibrium solubility of a compound starting from a solid sample and incorporating a 66-h shaking period followed by a double-centrifugation separation stage. This approach avoids the issues associated with filter plate techniques. The liquid handling robot along with bespoke integrated appliances, such as the decapper unit and centrifuge, offer a large degree of automation. This automation compensates for the increased experimental complexity and facilitates the greater throughput capacities of up to two plates of 48 compounds in one assay. With this technique, we are able to support drug discovery projects much earlier in the screening cascade with reliable aqueous equilibrium solubility data.
Footnotes
Acknowledgment
Competing Interests Statement: The authors certify that they have no relevant financial interests in this manuscript.