
Abstract
Preparative HPLC-MS is often the method of choice for purification of small amounts (<100 mg) of diverse new molecules, such as compound libraries for drug discovery. The method is robust, well proven, and widely applicable. In contrast, preparative supercritical fluid chromatography coupled with mass spectrometry (SFC-MS) has seen only slow acceptance for the same application—despite some potential scientific and economic advantages. One of the reasons for slow adoption of SFC-MS is the lack of well-proven, robust, and commercially available instrumentation. In early 2009, TharSFC (a Waters Company, Pittsburgh, PA) introduced a new fully integrated system for preparative SFC-MS: The SFC-MS Prep-100. We report herein an objective evaluation of the SFC-MS Prep-100, including tests for pump and autosampler performance, sample recovery, sample carryover, fraction triggering, detector/fraction collector synchronization, and overall robustness. Our results suggest that the SFC-MS Prep-100 represents a significant advance over previous generation instrumentation.
Keywords
Introduction
Mass-directed reverse phase gradient HPLC purification is the most commonly used technique for the purification of diverse small molecules in a high-throughput purification environment, 1,2 and a wide selection of suitable instruments is commercially available. Gradient preparative supercritical fluid chromatography (SFC) may offer potential advantages over HPLC, including nonaqueous purification, shorter run times, and higher efficiency because of low eluant viscosity and high solute diffusivity, 3 but it has not yet become widely accepted, and the selection of available instruments is limited.
Gradient preparative SFC systems with ultraviolet (UV) absorbance-directed fractionation and open-bed fraction collection were first introduced in year 2000, 4 and such systems have been successfully used for purification of combinatorial library compounds. 5 –8 Mass-directed fractionation is often preferred over UV-directed fractionation in the high-throughput diversity environment because it minimizes the need for downstream fraction analysis to identify the target peak. To address this need, the first commercially available gradient preparative SFC-MS system, the Prep-30, was introduced by TharSFC (a Waters Company, Pittsburgh, PA) in 2008. Since introduction, this instrument has been more fully described in the literature. 9,10 Key findings are that the Prep-30 provides reasonable (80%-85%) compound recoveries and minimal cross-contamination at flow rates up to 30 g/min. This maximum flow rate was viewed by many as too low for the target application, and in response, TharSFC introduced the SFC-MS Prep-100 system (Fig. 1), with a maximum flow rate of 100 g/min, in early 2009. The higher flow rate, in turn, increases the magnitude of some of the key instrument challenges, including gas expansion and resulting spray, cross-contamination, and detector/fraction collector synchronization. This article describes an evaluation of the new TharSFC SFC-MS Prep-100 purification system, with emphasis on the key challenges as previously highlighted.

TharSFC SFC-MS Prep-100 purification system. Reprinted with permission. Copyright TharSFC.
System Configuration
All preparative SFC-MS experiments were run on a TharSFC SFC-MS Prep-100 Preparative SFC-MS system equipped with Waters 2767 autosampler and autoinjector, Waters 3100 MS Detector, Waters 2998 photodiode array (PDA) detector, Waters 515 pumps, TharSFC high-pressure CO2 and solvent pumps, 6-port column-switching column oven, TharSFC preparative automated back pressure regulator, TharSFC tunable splitter, TharSFC gas-liquid separator (GLS), and controlled by Waters MassLynx 4.1 SCN 627 software (Waters Corporation, Milford, MA). Unless otherwise noted, all mass spectra were acquired using the atmospheric pressure chemical ionization mode, with cone voltage of 40 V and probe temperature set at 500 °C. Unless noted, chromatograms were run on a 21.2 × 100-mm 2-ethylpyridine (2-EP) (5 μm) column from Princeton Chromatography (Princeton, NJ) using 100-g/min total flow. Unless otherwise noted, the organic cosolvent was 10-mM ammonium acetate in methanol (MeOH). A general system schematic is shown in Figure 2.
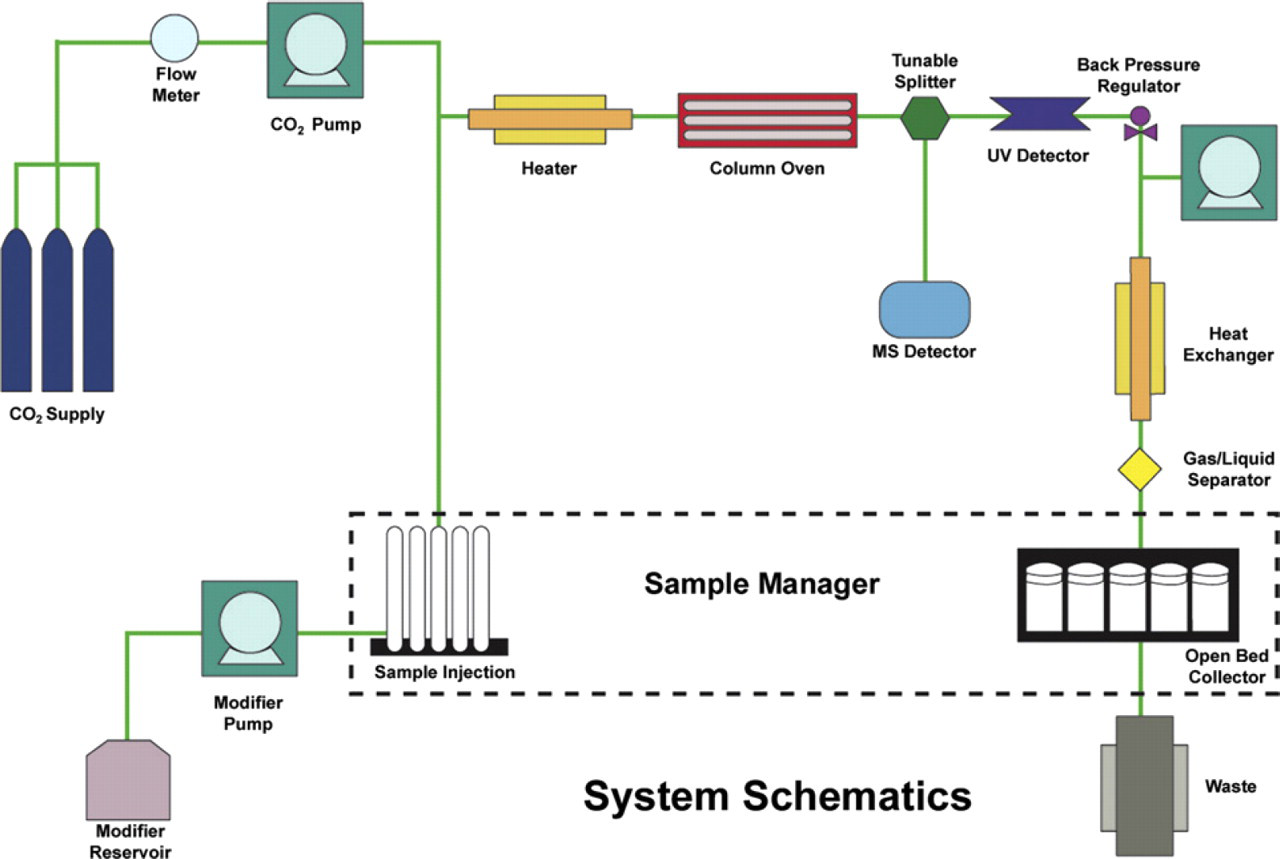
Schematic of TharSFC SFC-MS Prep-100 SFC-MS system. Reprinted with permission. Copyright TharSFC.
Evaluation of PUMP and Mixing Performance
Gradient chromatography requires a system with relatively low extra-column volume to minimize band tailing and broadening, a dwell volume as small as possible to reduce the gradient delay, 11 accurate proportioning by the solvent delivery system, and efficient mobile phase/analyte mixing. To evaluate the efficiency of solvent mixing, the column was replaced with a zero dead-volume union, and a linear gradient was run (Table 1) using MeOH containing 5% acetone as the organic cosolvent. UV absorbance was measured at 254 nm, and the result is shown in Figure 3. We observed an error-free gradient, with no dispersion or deviation from the linear line. The system dwell volume (V D) was calculated by using V D = T d × F, where T d is the dwell time and F is the flow rate (100 mL/min). The observed T d is 0.5 min; thus, the calculated V D of the system is 50 mL (Fig. 3).
Gradient linearity test method

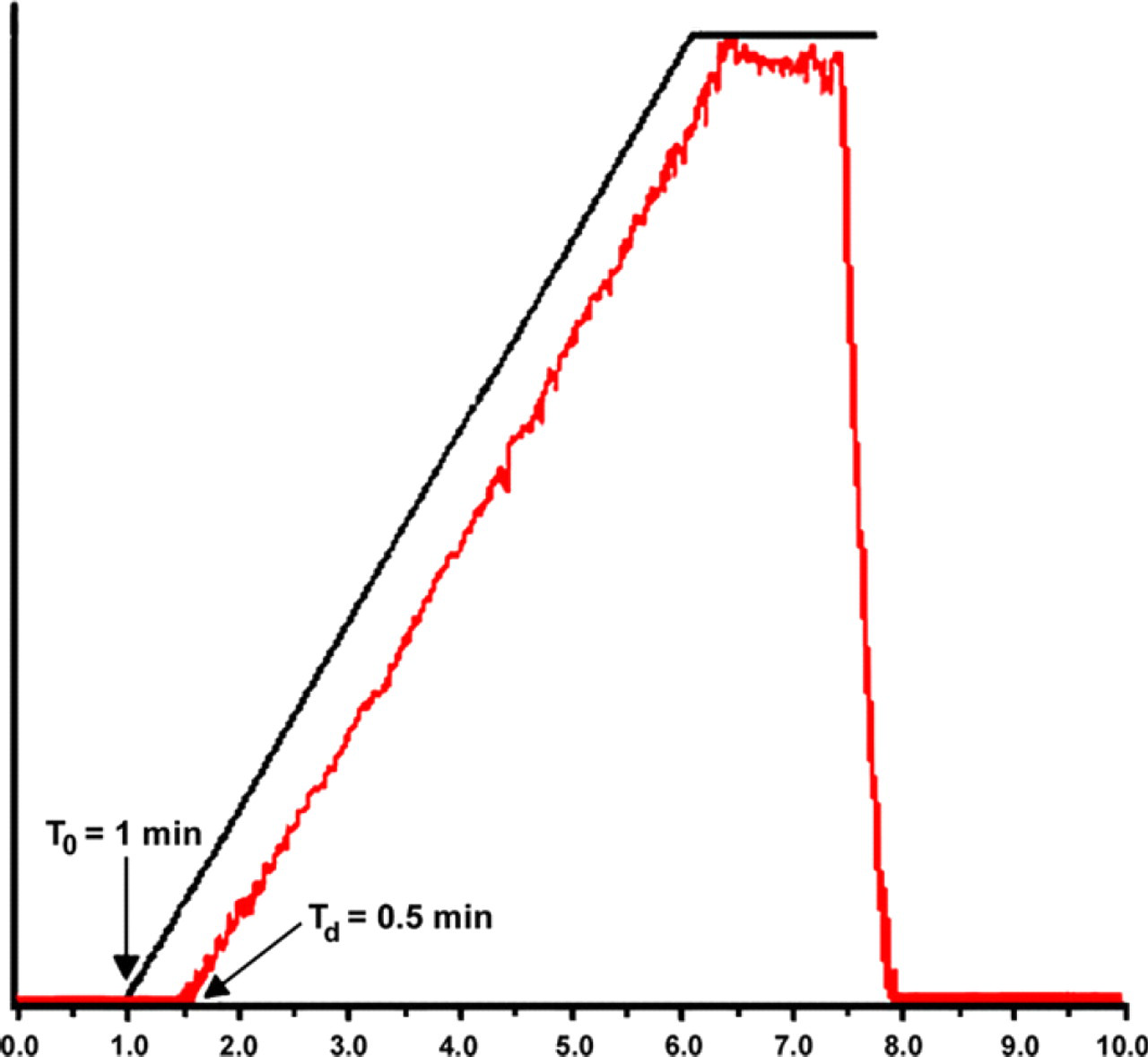
Gradient linearity test results.
The accuracy of solvent proportioning was investigated by using a step gradient consisting of 10% steps, each held for 2 min (Table 2). The chromatogram of this experiment was recorded at 254 nm and is shown in Figure 4. The distance between each plateau is identical and equal to 10% of the full-scale response (standard deviation less than 1%), indicating even proportioning through the gradient range.
Gradient proportioning test method

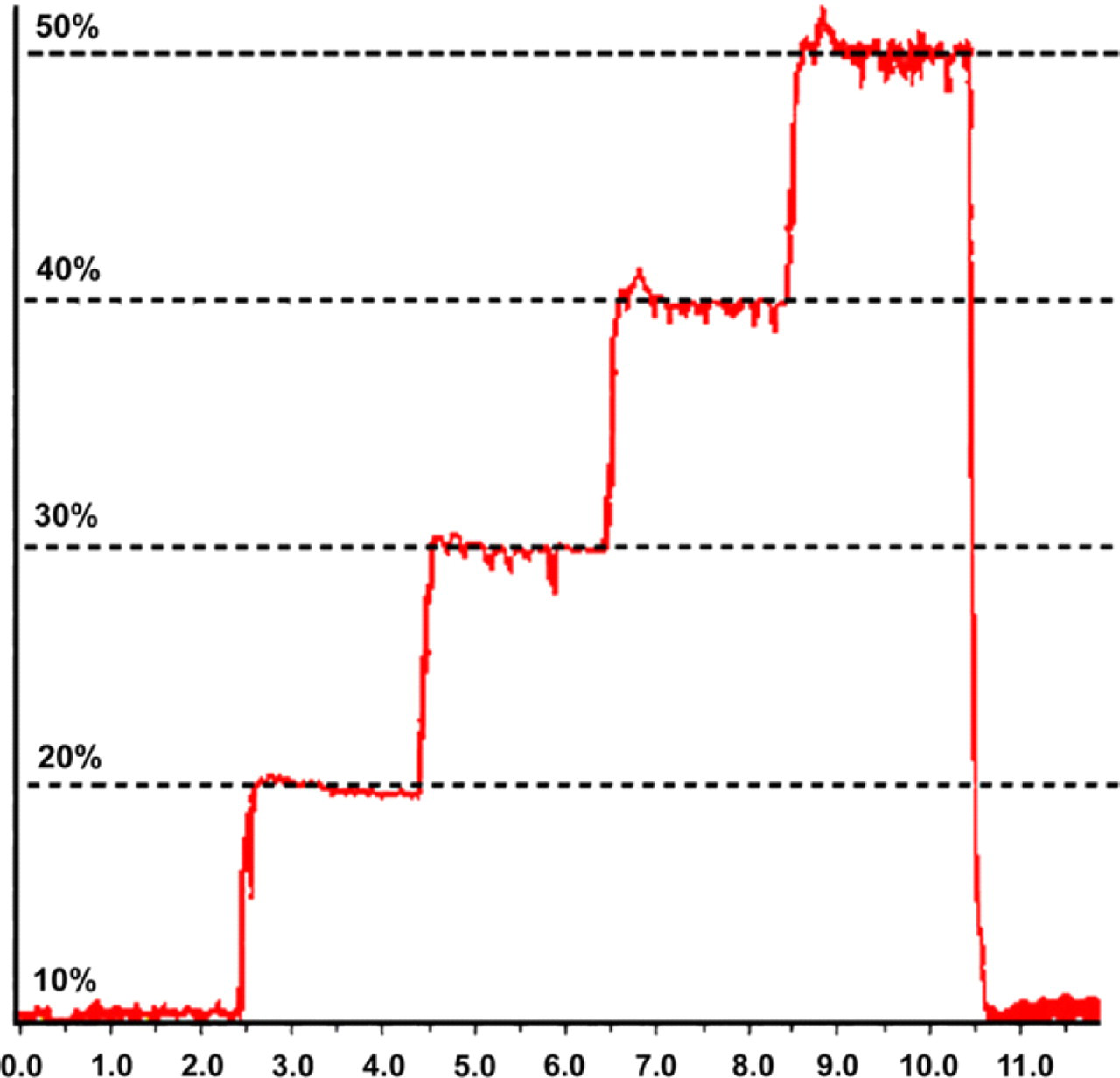
Gradient proportioning test results.
Evaluation of Autosampler Performance
The most important autosampler performance features related to preparative purification are overall robustness, aversion to clogging, and z-axis precision (to ensure sampling and injecting the entire sample). Although accuracy and precision of injection volume are less critical in day-to-day operation of preparative systems, some of the tests we performed as part of this evaluation do rely on injection volume reproducibility. This was thus measured by performing 10 consecutive 1.0-mL injections from test solutions containing either flavone (41.4 mM in dimethylformamide [DMF]) or sulconazole (34.7 mM in DMF) onto a 2-EP column, 12 followed by gradient elution using the gradient described in Table 1. UV absorbance was measured at either 320 nm (flavone) or 235 nm (sulconazole); peaks were integrated and total peak area was measured for each injection. Standard deviation for peak areas (from 10 injections) was 0.34% for flavone and 0.17% for sulconazole, suggesting very good injection-to-injection reproducibility.
Evaluation of System Loading
One of the primary target applications for the Prep-100 system is purification of “first lot” pharmaceutical compounds from drug discovery. In this application, samples sizes are typically less than about 100 μmol but occasionally range up to several hundred micromoles. Typical stationary phases used for this application include 2-EP and other bonded silica phases, 5,12 and typical configurations are 20-mm diameter × 100–250-mm length. Although loading is a function of column chemistry, it is also a function of solvent and sample mixing and delivery. 13 We thus measured the impact of both mass and volume 11 overload on overall separation performance using the 2-EP column.
For the mass overload study, 1-mL injections containing increasing concentrations of either flavone or sulconazole (ranging from ∼ 10 to ∼400 mM in DMF) were injected onto the 2-EP column, followed by gradient elution, using the gradient described in Table 1. UV absorbance was measured across the full spectrum using the PDA detector. Absorbance linearity (total peak area vs amount injected) was later measured using various extracted wavelengths, and subsequent data analyses were done at either 340 nm (flavone) or 280 nm (sulconazole), both of which showed high linearity (R > 0.99).
The goal of preparative purification is to achieve separation of the target compound from impurities. Separation is, in turn, defined by resolution, which is inversely proportional to peak width. 11 To estimate the potential impact of loading on resolution using only a single component, we measured peak width versus load and then plotted the ratio Widthmin/Widthload versus sample load (where Widthmin is the half-height peak width at lowest load and Widthload is the half-height peak width at the measured load). This plot approximates the anticipated falloff 14 in resolution of this compound from a similarly behaving compound as a function of load. Results for flavone are shown in Figure 5A and B, and results for sulconazole are shown in Figure 6A and B.
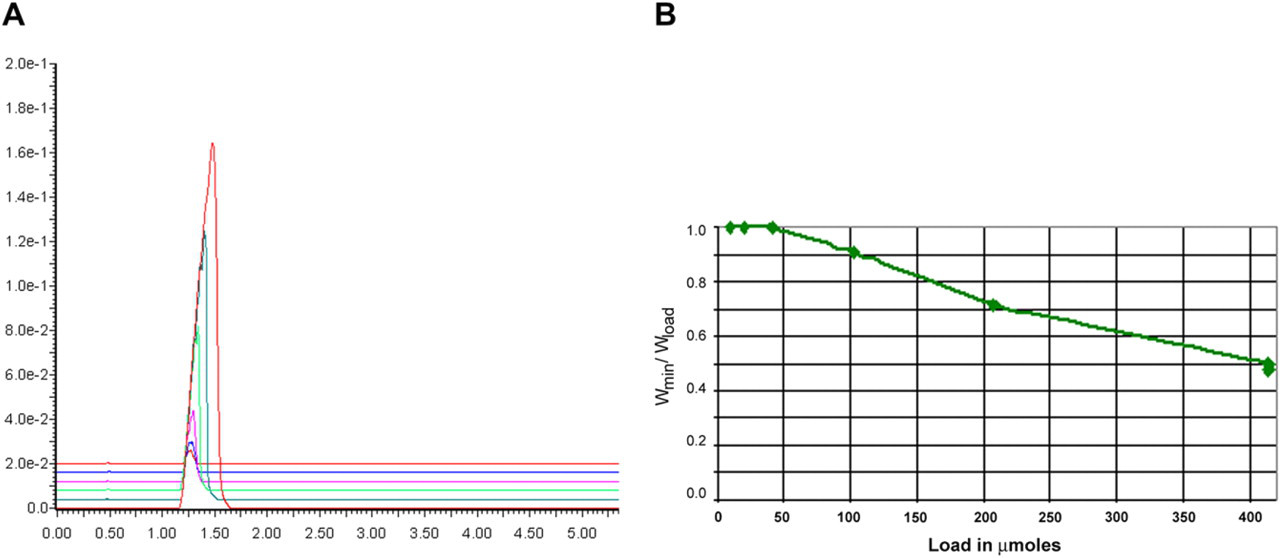
Mass overload results. (A) Overlaid chromatograms of flavone. (B) Impact of load on peak width ratio of flavone.
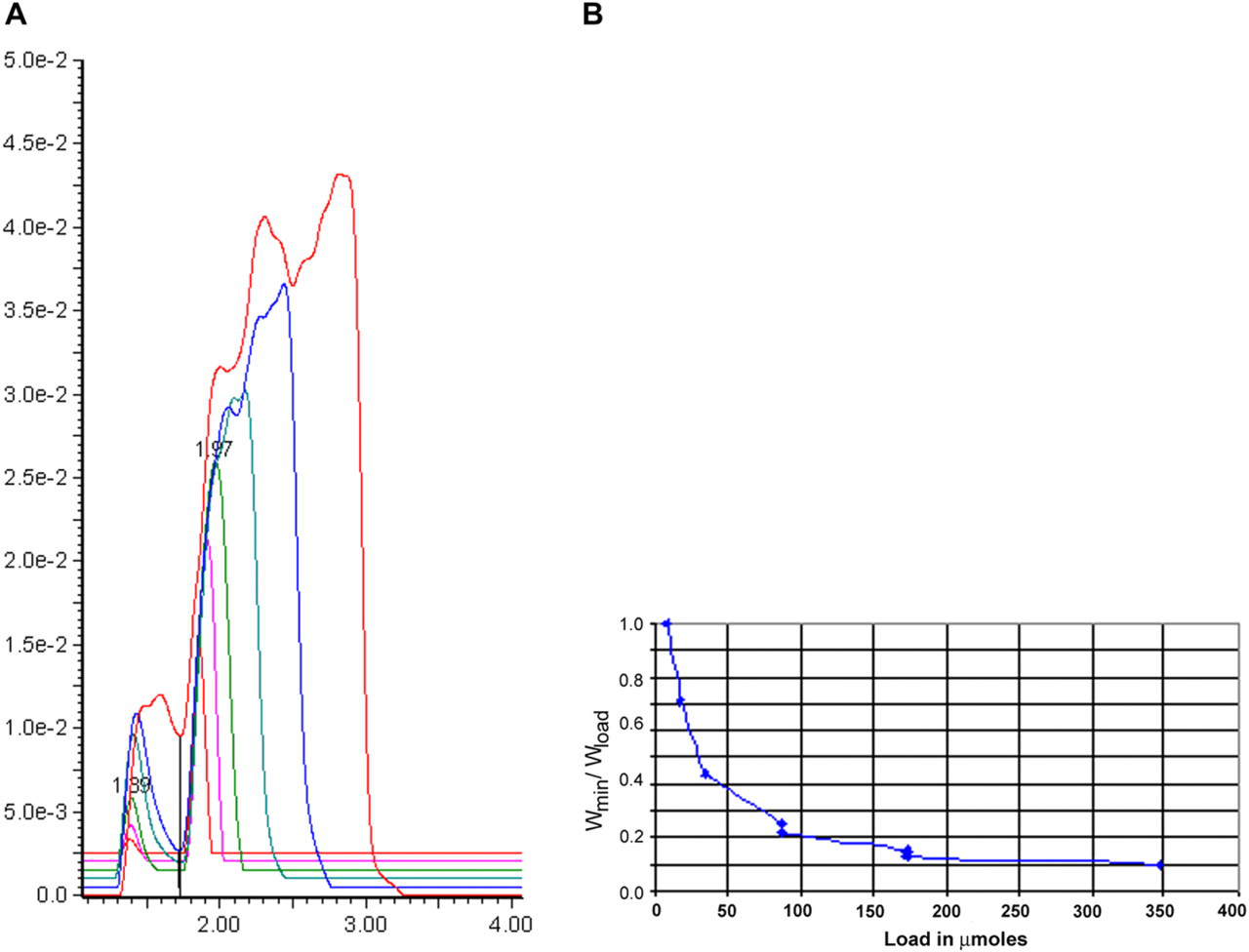
Mass overload results. (A) Overlaid chromatograms of sulconazole. (B) Impact of load on peak width ratio of sulconazole.
As shown in Figure 5A and B, flavone (a neutral compound) retains reasonable peak shape throughout the loading range and is predicted to lose about half the resolution at the highest load. This result is qualitatively similar to results observed for flavone under typical preparative HPLC conditions (not shown). Importantly, it shows that there are no unexpected injection or mixing effects as the load increases. Sulconazole, a basic compound, behaves quite differently (Fig. 6A and B). Figure 6A shows overlaid chromatograms at increasing sulconazole load (a small DMF peak is observed at about 1.4 min in the sulconazole traces because of the wavelength monitored). The main sulconazole peak at about 2.0 min retains reasonable shape up to a load of 35 μmol (trace 3 of 6); above that load, the peak begins to fragment into multiple partly resolved peaks. We believe that this may be because of a weak buffering effect: the MeOH contains 10-mM ammonium acetate, but the starting gradient contains only 5% MeOH in the mobile phase, thus only 50 μmol of the buffer are delivered in the first minute of the gradient. This differs from HPLC where it is possible to introduce buffer into both weak and strong mobile phase components, whereas for SFC, buffers can only be introduced via the modifier solvent (in this case MeOH). Further studies will be needed to completely understand buffering in preparative SFC.
Volume overload was measured by injecting increasing volumes of analyte solution (in DMF), each containing the same total amount of analyte. Total analyte amounts were 41 and 35 μmol, respectively, for flavone and sulconazole—both of which were within the limit of reasonable peak shape in the mass overload study previously described. The column and gradient method was the same as previously described for the mass overload study. Injection volumes ranged from 0.1 to 2.0 mL. Results are shown in Figure 7A (flavone) and B (sulconazole). As shown in the figures, peak shapes remain reasonable throughout the volume range and peak areas remain constant (standard deviation 3% for flavone and 2% for sulconazole).
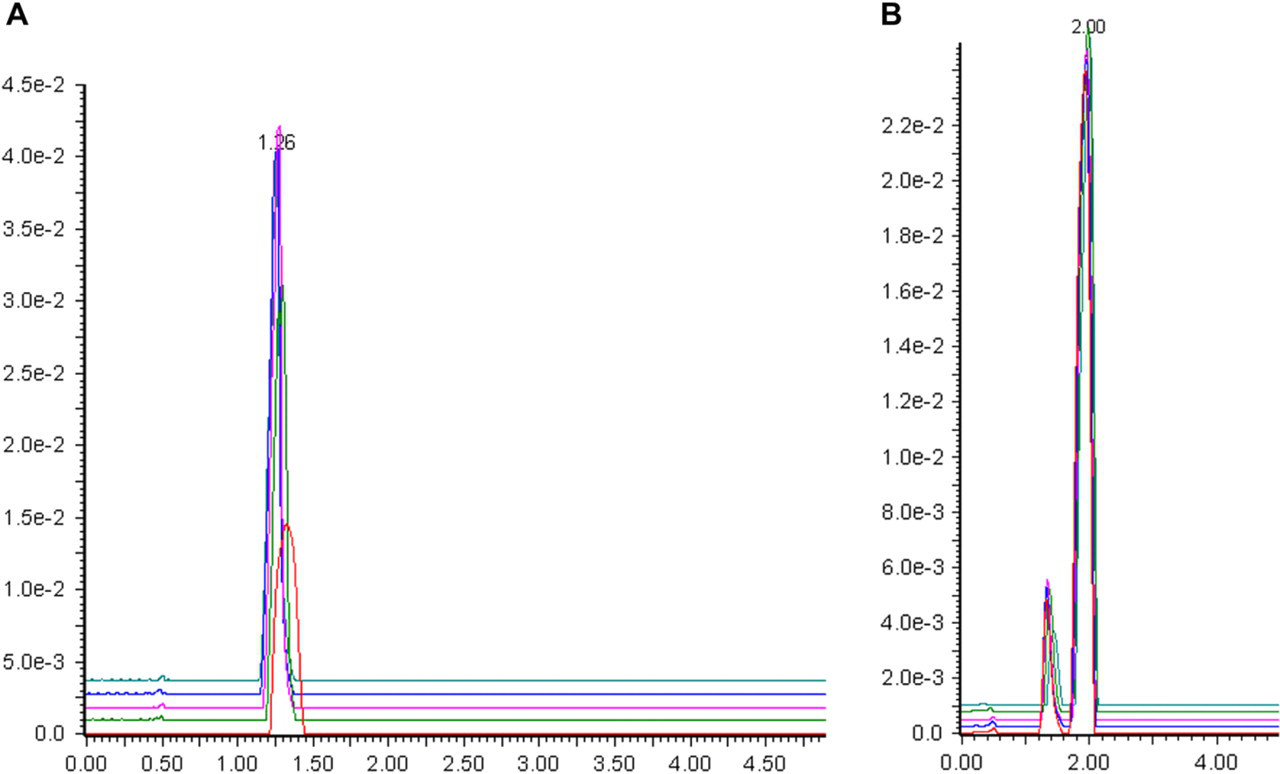
Volume overload results. (A) Flavone. (B) Sulconazole.
Evaluation of Fraction Triggering
Fraction triggering on the system is controlled by the Waters FractionLynx software 15 with parameters set via the standard FractionLynx interface. Triggering options include UV at specified wavelength, MS-extracted ion chromatogram as specified mass, or Boolean logic combining both detection methods.
In preparative SFC, rapid gas expansion occurs downstream of the detector and before the fraction collector. This expansion is very rapid and can cause extensive spraying and mist formation, which might lead to loss of sample or sample cross-contamination. In bulk SFC purification systems, this is addressed by use of a gas liquid separator (GLS) that allows gas to escape while drawing the liquid eluant to the bottom of a large glass tube. An example of such a separator is shown in Figure 8. 16 This system is effective in bulk purification, where a large amount of compound is purified by repeated injection of smaller amounts of the same sample. In this mode, the next injection effectively washes down the residue from the first injection, and so forth, so that the bulk of the sample is collected after repeated injections. Any mist that is lost because of gas expansion contains exactly the same material as the next injection, so cross-contamination by washing back of the mist into the sample tube is not an issue. This configuration, however, is less desirable in an application where each injection contains a unique sample. In this case, the volumes and flushing time required to adequately rinse the concentrator could be unacceptably high.
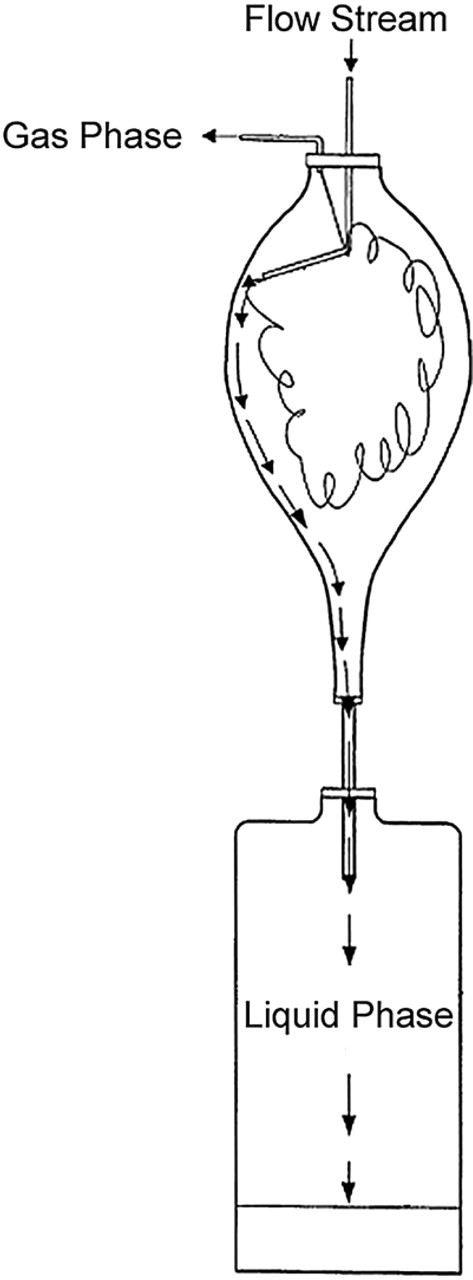
Typical GLS for bulk purification. 16
Previous generation SFC purification systems for diverse compounds using flat-bed collection eliminated the GLS entirely and replaced it with a deflector that guides the spray toward the bottom of an overly large collection tube. 6,17,18 Although these systems have proven to be effective, 9 they have limitations, including the use of an overly large collection tube and uncontrolled misting that holds potential for sample loss or contamination. Additionally, this mechanism only works at relatively modest flow rates—for example up to 30 mL/min with the TharSFC Prep-30 system. To enable the higher flow rates of the SFC-MS Prep-100 (100 mL/min), TharSFC designed a novel GLS. The SFC-MS Prep-100 system uses a new proprietary GLS, which allows minimal interaction between the gaseous CO2 and the solvent while maintaining flow stream integrity so that gas can be vented and solvent with dissolved compound can be collected under ambient pressure. In these experiments, a 5 s rinse time was set to rinse the collection probe between fractions. Because the GLS is proprietary, it has not yet been fully described; our tests were thus designed to test the functionality of the system without concern as to its design.
To evaluate fraction triggering, cross-contamination, and sample recovery, we injected a dimethylsulfoxide (DMSO) solution (1.5 mL) containing three components: caffeine (68 μmol, early eluting), sulconazole (81 μmol, middle eluting), and bendroflumethazide (178 μmol, late eluting) onto the 2-EP column and eluted using the linear gradient method in Table 1, except with the gradient time extended from 6 to 7 min. Fractions were collected using each of the three triggering methods: UV only (220 nm), MS only (M+H)+, or UV and MS Boolean; each using the standard FractionLynx Auto MIT method to set the minimum intensity threshold. All injections were made in triplicate. Collected fractions were dried on a GeneVac HT 24 evaporator (Genevac LTD, Gardiner, NY) (using two full drying cycles of 16 h at 45 °C and 0.4 mbar) and weighed. Average recoveries in percentage (Pct recovery) are shown in Table 3; standard deviations in all cases were small (<0.5%).
Sample Recovery
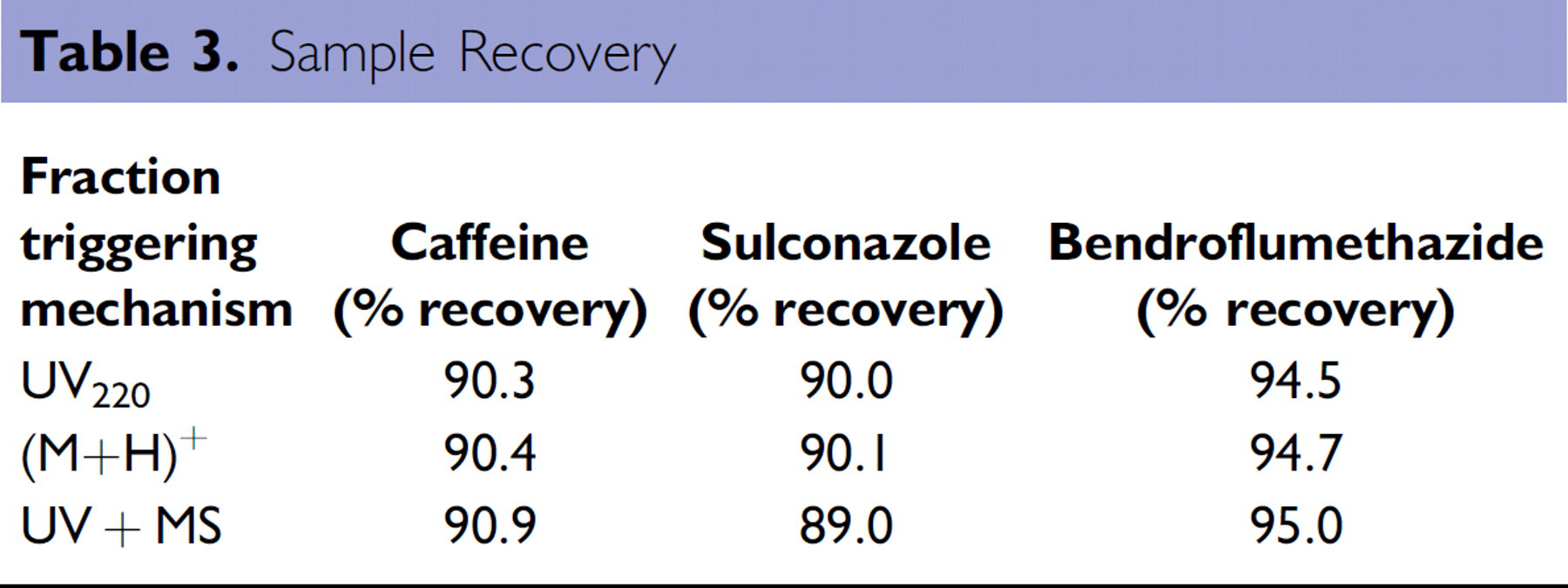
As seen in the Table, recoveries were high (∼90%+) for all three fraction triggering methods. This stands in contrast with a similar experiment we performed previously on an earlier generation instrument (the TharSFC Prep-30), lacking a GLS and using an open-bed fraction collector, where maximum recoveries were in the range of 80%. 19 This demonstrates the effectiveness of the new GLS in reducing sample loss because of misting.
Recovered fractions were analyzed for purity by gradient analytical LC-MS. 20 In all cases, purity (area percent by UV at 220 nm) was greater than 99%, and, in particular, none of the other two mixture components was observed as a contaminant in any of the recovered products to the limit of detection (<0.1%). Once again, this stands in contrast to our previous results with the earlier Prep-30, where we observed cross-contamination (compound from peak N contaminating compound from peak N + 1) in the range of 2%-3%. 19
The observed high purity and recovery suggest that new GLS is doing its job well and that the various detectors are properly synchronized. The slightly higher recovery observed for the late eluting component is consistent with our previous observations with earlier preparative SFC systems and may be because of the higher percentage of cosolvent (MeOH), and correspondingly lower percentage of gas released, late in the gradient.
As with all preparative chromatography systems, delay times between the detector(s) and the fraction collector head must be taken into account to ensure proper synchronization. In the case of SFC, gas expansion takes place immediately downstream of the detector and before the fraction collector (Fig. 2)—thus the flow rate between detector and fraction collector will be vary as the gradient composition changes. In addition, the postsplit flow rate could vary if the split ratio itself changes in response to pressure or viscosity changes. These factors create synchronization challenges that are unique to SFC. The SFC-MS Prep-100 system addresses this challenge by addition of a makeup pump immediately after gas release. This pumps an approximate reverse gradient, the exact composition of which is determined by a proprietary algorithm that takes into account system volume delay, to ensure approximately constant liquid flow through the system between gas release and fraction collection.
To assess the effectiveness of the makeup pump in maintaining synchronization, the three-component sample used in the recovery test previously outlined was injected onto the same column as before and eluted using the same gradient method. This time, however, fractions were collected continuously every 6 s from 0.5 to 6.5 min (the interfraction wash was turned off for this experiment). After collection, all 60 collected fractions were dried, reconstituted to constant volume using 1 mL of DMF, and analyzed by analytical SFC-MS. 21
The purpose of the experiment was to verify constant alignment among chromatograms across the entire gradient. Chromatograms, including the reconstituted chromatogram from fraction analysis, are plotted in Figure 9. As seen from the figure, the alignment was constant throughout the entire gradient: caffeine was found in fractions 5–10 (vs predicted from chromatogram: 5–10), sulconazole was found in fractions 14–24 (predicted: 14–24), and bendroflumethazide was found in fractions 44–51 (predicted: 44–51). The alignment between detector-based chromatograms and fractions actually collected remained constant throughout the gradient; thus, the makeup pump performed as anticipated in maintaining alignment between detectors and fraction collector. This, in turn, contributed to the high recoveries observed in the previous experiment.
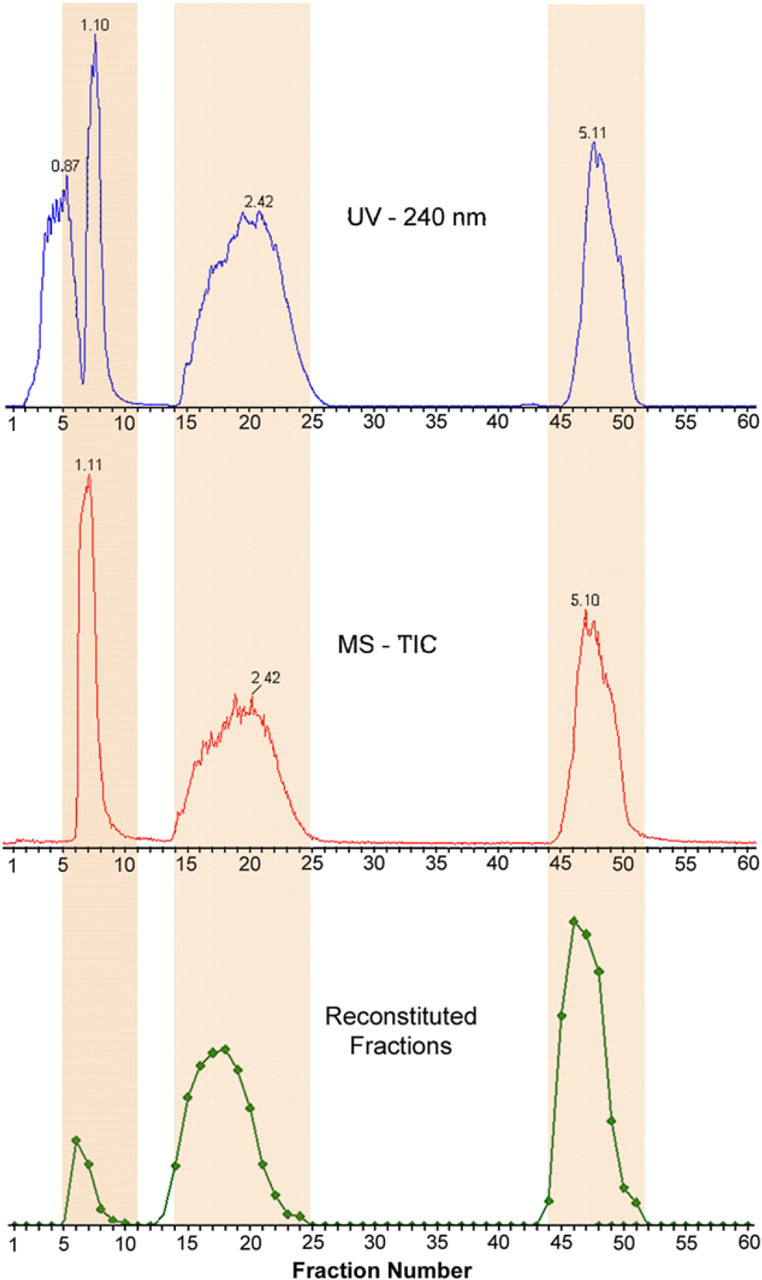
Peak alignment throughout gradient elution.
Full System Test
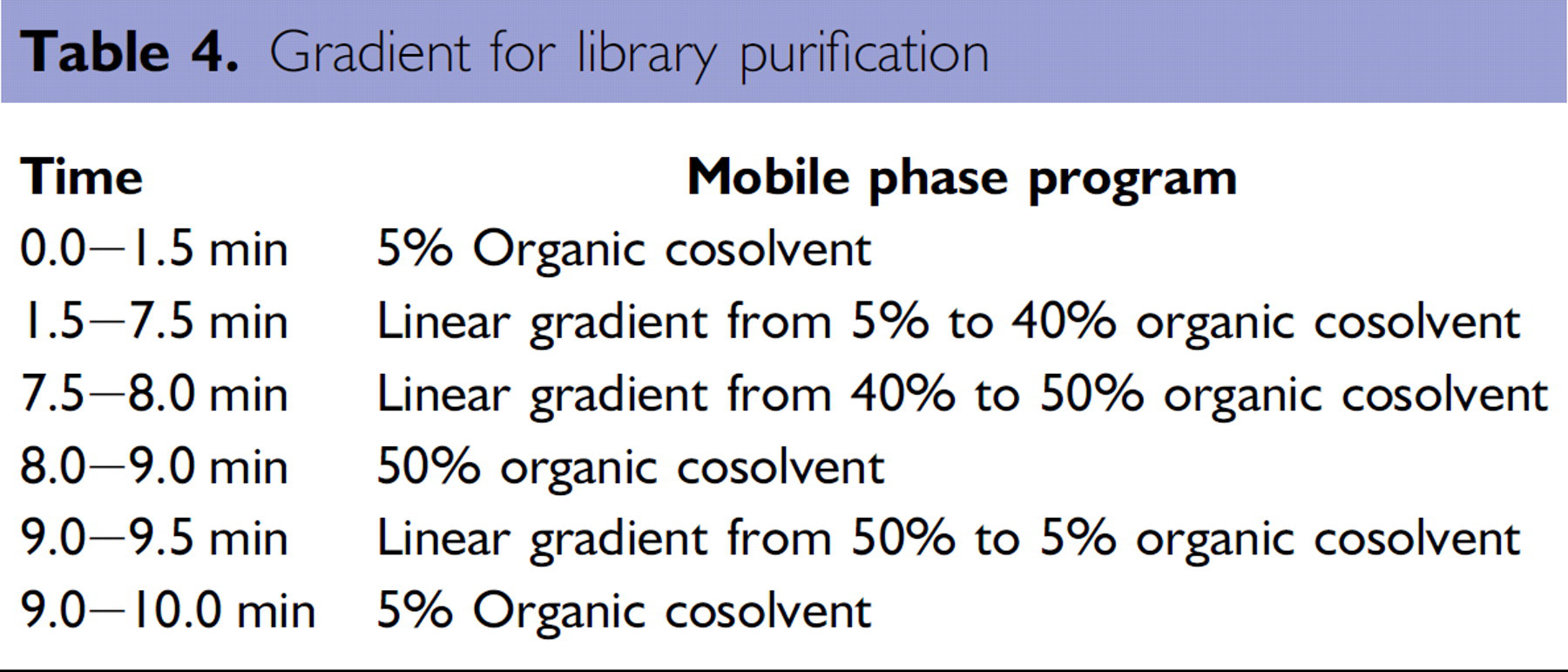
To evaluate the effectiveness of the system for its intended application, we purified two small (24-member) synthetic compound libraries. Chemical reactions were run at 60-μmol scale. The first library was from a simple amidation reaction and had starting purities (target area percent) ranging from 11% to 65% (average 48%) by analytical SFC-MS with detection by UV at 220 nm. The crude reaction products were dissolved in 1.5 mL of DMSO and were purified on the 2-EP column using the gradient in Table 4, with 10-mM ammonium acetate in MeOH as cosolvent. The same generic gradient was used for all samples, and the method was not optimized for specific compounds or for these specific libraries of compounds.
Gradient for library purification
Fractions were collected by MS triggering on the (M+H)+ ion as described in the previous section. Fractions were dried, weighed, and analyzed by SFC-MS to determine the purity (UV area percent). Average purity was 98%, with 23 out of 24 products having purity >95% (the 24th was 90% pure). Isolated chemical yield ranged from 5% to 83% (average 42%). This library was synthesized in duplicate, and the companion library was purified by traditional preparative HPLC-MS. Average purity was also 98%, with 23 out of 24 products having purity >95% (the 24th was 85% pure); average isolated chemical yield was 56% (13%–83%). Purification by SFC and HPLC thus gave similar results for this library. The small difference in isolated chemical yield may be an artifact of shipping and sample handling: the SFC samples were shipped to another site for dry down and analysis, whereas the HPLC samples were processed locally. A second library, also 24 compounds at 60-μmol scale from a more complex Suzuki-type coupling reaction, had initial purity ranging from 4% to 65% (average 58%) and final purity ranging from 80% to 100% (average 95%); average isolated chemical yield was 42%. In all cases, the purifications were run continuously and unattended and proceeded without failure. Although it will take a much larger sample size to determine overall system suitability and robustness, these initial experiments suggest a positive outcome.
Summary
Widespread adoption of preparative SFC for purification of diverse small molecules has been hampered by the lack of suitable commercially available instrumentation capable of providing reasonable flow rates, gradient elution, mass-driven fractionation, and consistent high recoveries without cross-contamination. Key technical hurdles included providing adequate sample and solvent mixing throughout the gradient, managing gas expansion to avoid sample loss or contamination, and managing detector/fraction collector synchronization in gradient elution. The experiments previously outlined suggest that the SFC-MS Prep-100 has successfully addressed each of these potential issues. As these systems begin to be deployed into working laboratories, system robustness and suitability for the target application will become fully tested. If the instrument ultimately performs to its full potential, it may, for the first time, allow evaluation of the underlying scientific and commercial value of preparative SFC relative to HPLC without being hampered by underlying instrumentation limitations.
Acknowledgments
The authors thank TharSFC for providing extended instrument access and in particular Dr. Rui Chen and Ms. Lakshmi Subbarao for their technical assistance during the experiments. They thank Dr. David Nirschl (Bristol—Myers Squibb [BMS]) for synthesizing the compound libraries used for this evaluation, members of the BMS Discovery Analytical Sciences department and the Synthesis and Analysis Technologies department for ongoing technical discussions on the topic of SFC purification, and the management of the BMS Discovery Chemistry and Applied Biotechnology departments for overall project support.
Competing Interests Statement: The authors certify that all financial and material support for this research and work are clearly identified in the manuscript.