
Abstract
We describe a new approach to the sensitive and specific identification of bacteria, viruses, fungi, and protozoa based on broad-range PCR and high-performance mass spectrometry. The Ibis T5000 is based on technology developed for the Department of Defense known as T.I.G.E.R. (Triangulation Identification for the Genetic Evaluation of Risks) for pathogen surveillance. The technology uses mass spectrometry—derived base composition signatures obtained from PCR amplification of broadly conserved regions of the pathogen genomes to identify most organisms present in a sample. The process of sample analysis has been automated using a combination of commercially available and custom instrumentation. A software system known as T-Track manages the sample flow, signal analysis, and data interpretation and provides simplified result reports to the user. No specialized expertise is required to use the instrumentation. In addition to pathogen surveillance, the Ibis T5000 is being applied to reducing health care—associated infections (HAIs), emerging and pandemic disease surveillance, human forensics analysis, and pharmaceutical product and food safety, and will be used eventually in human infectious disease diagnosis. In this review, we describe the automated Ibis T5000 instrument and provide examples of how it is used in HAI control.

Introduction
The Ibis T5000 universal biosensor is a unique technological approach to infectious agent identification. The system emerged from technology developed under a Defense Advanced Research Projects Agency (DARPA) program known as T.I.G.E.R. (Triangulation Identification for the Genetic Evaluation of Risks) 1 , originally developed for biological weapons defense. In its commercial form, the Ibis T5000 is a universal pathogen detection platform capable of identification and strain typing of a broad range of pathogens 2, 3 (Fig. 1). The fundamental difference between the Ibis T5000 universal biosensor and existing methodologies is the nature of the question being asked. Current infectious organism detection systems answer specific questions of the form “Is a certain pathogen present in my sample?” The Ibis T5000 answers the question, “What infectious organism(s) are in my sample?” In effect, use of the Ibis T5000 is equivalent to running many thousands of specific identification reactions because the identity of the infectious organism does not need to be anticipated. The platform is also capable of providing additional information about the microbe such as its genotypic fingerprint, whether it is resistant to certain antibiotics, and whether it carries certain virulence factors. The technology is currently being developed and used for pathogen surveillance, health care–associated infection (HAI) control, public health epidemiology, and human and animal clinical research.

The Ibis T5000 universal biosensor. Shown in this view are key modules including amplicon purification and the mass spectrometer. Precise molecular weight determinations of amplicons yield unambiguous base compositions that are used to uniquely “fingerprint” each pathogen. The automated system is capable of analyzing over 1500 PCRs in 24 h.
This approach is enabled by, first, the use of broad-range primers to amplify PCR products from groupings of organisms, rather than single organisms, and second, the use of mass spectrometry to determine the base compositions of the products. Unlike nucleic acid probes or arrays, mass spectrometry does not require anticipation of products analyzed, but simply measures the masses of the nucleic acids present in the sample. The analog signal of mass is converted to a digital signal of base composition based on the accuracy of the mass measurement and the discrete masses associated with different combinations of the four nucleotide bases. Mass spectrometry is remarkably sensitive and can measure the mass and determine the base composition from small quantities of nucleic acids in a complex mixture with a throughput exceeding one sample per minute. The ability to detect and determine the base composition of a large number of PCR amplicons in a mixed sample enables analysis and identification of PCR products essentially instantaneously. It is broadly accepted that the nucleic acid sequence of specific regions of a genome can be used to identify and differentiate pathogens. Although not as intuitive, nucleic acid compositions (i.e., the number of A's, G's, C's, and T's) in specific regions of a genome are equally informative and can be derived in a fully automated high-throughput modality.
Principles of Operation
The T.I.G.E.R. process is illustrated in Figure 2 and is summarized briefly below. We recently published a detailed description of the technology. 2 The process begins (Fig. 2, Step #1) with the extraction of all nucleic acids present in a sample. The sample is aliquoted into wells of a microtiter plate that each contains one or more pairs of broad-range primers for PCR. The broad-range primers are designed to amplify a product from a group of organisms from a selected domain of microbial life, for example, all bacteria or specific bacterial divisions. The PCRs produce a mixture of products reflecting the complexity of the original mixture of organisms present in the starting sample.
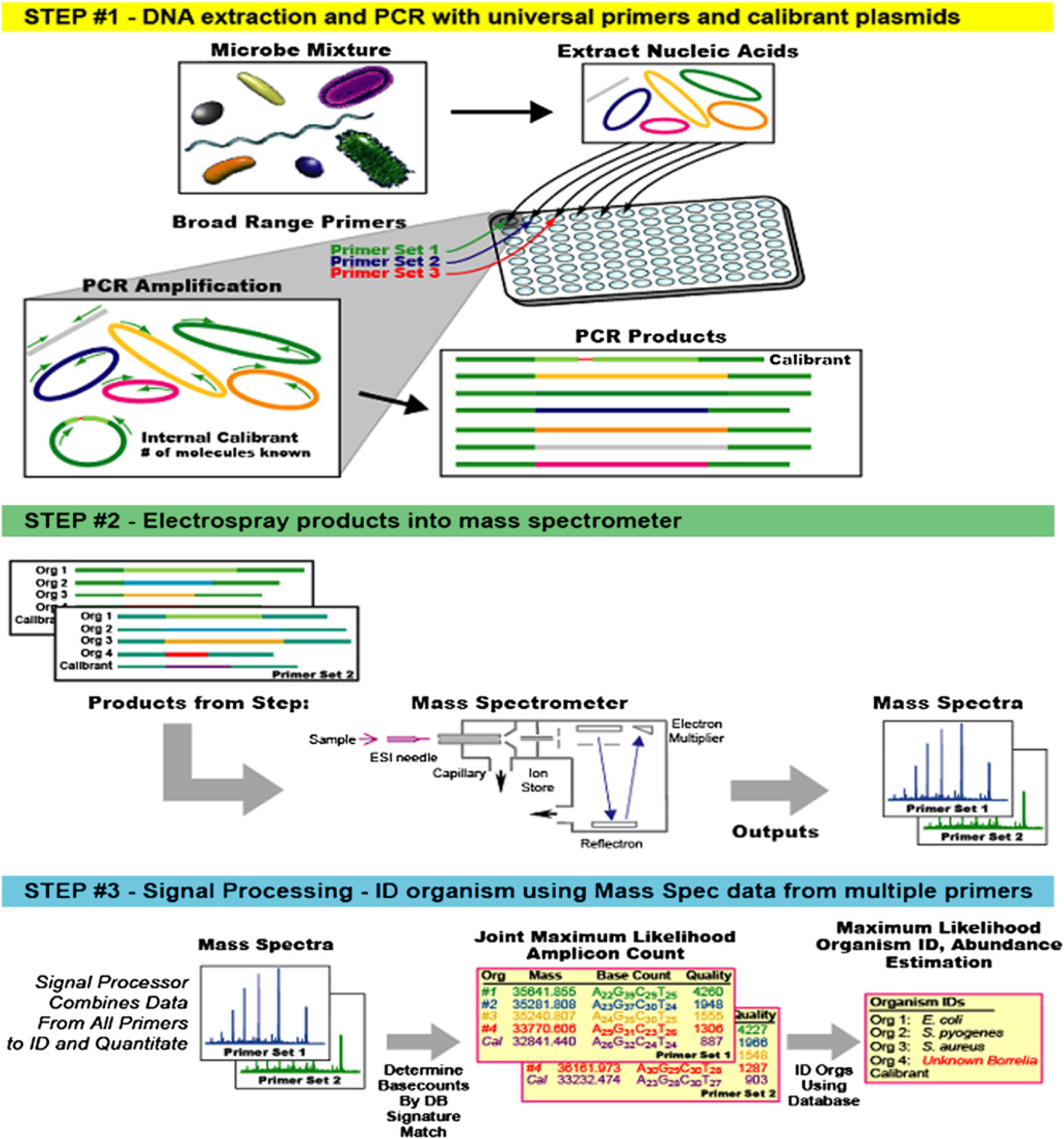
The T.I.G.E.R. concept of operation. In Step #1, nucleic acid extracts from the sample of interest (e.g., air sample, clinical specimen, food product) are extracted and amplified with broad-range PCR primers. Step #2 is mass spectrometry–based analysis of PCR-derived amplicons. Signal processing in Step #3 yields unambiguous base composition signatures from multiple genomic regions that are in turn used to identify the microbe(s) in the sample.
The products from the PCRs are desalted in a 96-well plate format 4 and sequentially electrosprayed into a mass spectrometer (Fig. 2, Step #2). The spectral signals are processed to determine the masses of each of the PCR products present with sufficient accuracy that the base composition of each amplicon can be unambiguously deduced (Fig. 2, Step #3). Using combined base compositions from multiple PCRs, the identities of the pathogens and their relative concentrations in the starting sample can be determined.
The T.I.G.E.R. method is based on the principle that, despite the enormous diversity of microbes, all forms of life on earth share sets of essential common features encoded in their genomes. Bacteria, for example, have highly conserved sequences in universally conserved regions of the ribosomal RNA and in other noncoding RNAs, including RNAse P and the signal recognition particle among others. There are also conserved motifs in essential protein–encoding genes. These common, conserved features are used as anchors for broad-range PCR priming to generate amplicons from all organisms in an environmental or clinical sample without prejudice. The trade-off in broad-range priming compared with specific PCR is that PCR is a zero-sum game. The total yield of amplified product has an upper limit value that must be divided among all the targets amplified. It is essential that the technology detects the signal from the threatening or pathogenic organism in the background of an excess of harmless organisms. Although cloning and exhaustively sequencing many colonies can accomplish this, this method cannot be automated in a rapid diagnostic device. Our strategic breakthrough was the use of mass spectrometry to analyze the products of broad-range PCR. Mass spectrometry is remarkably sensitive and can measure the weight and determine the base composition from small quantities of nucleic acids in a complex mixture. We have demonstrated that such analyses are feasible in a high-throughput modality after a rigorous desalting treatment. 4 The ability to detect and determine the base composition of a large number of PCR amplicons in a mixed sample enables analysis and identification of broad-range PCR products essentially instantaneously.
Although the concept that base composition has resolving power is counterintuitive (one might suspect that sequences from different organisms will degenerate to similar overlapping compositions), a rigorous mathematical analysis has shown that composition retains resolving power that is more than sufficient to identify pathogens of interest. The mass accuracy provided by the mass spectrometer limits the base composition of each DNA strand to a finite number of possibilities. For example, Figure 3 shows mass spectra of PCR products derived from a sample containing Bacillus anthracis and an internal calibrant (amplified during PCR of the sample from an added plasmid) that allows calculation of the relative amounts of each organism present.
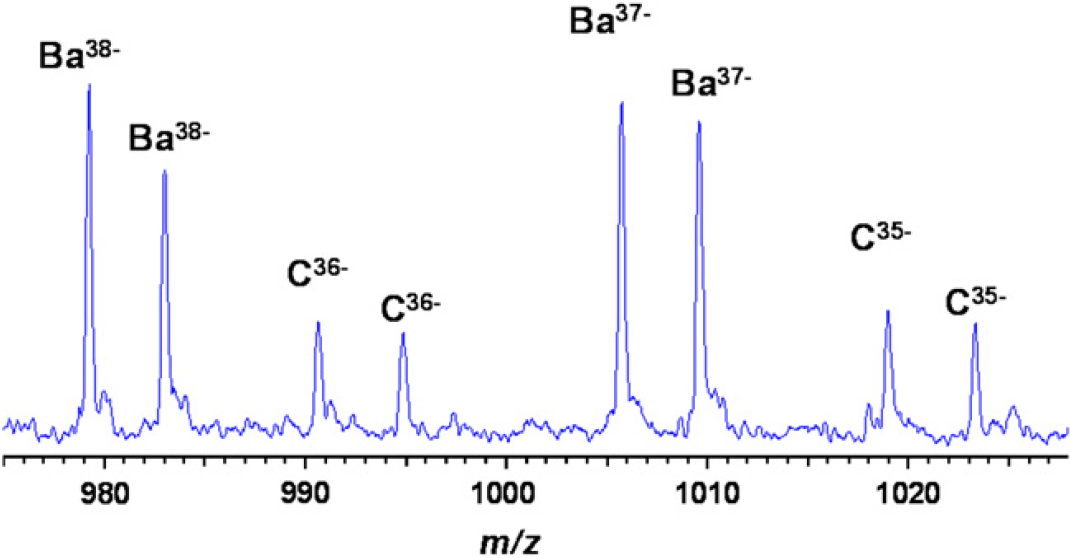
T5000 ESI-MS spectrum of PCR products derived from 1500 copies of B. anthracis in the presence of 100 copies of the plasmid calibrant using a broad-range primer that targets ribosomal protein L2 (rplB). Accurate mass measurements of the complementary strands allow unambiguous base composition determination of the B. anthracis amplicon (Ba). The internal calibrant (C) is used to calculate the relative amounts of each product from the PCR.
Table 1 lists the number of base compositions consistent with determined molecular weights (from Fig. 3) within a range of mass measurement uncertainties from 1 to 100 ppm. A mass measurement of either strand by itself, even at 1-ppm mass measurement error, is consistent with more than 100 base compositions for each strand, whereas a 20-ppm mass measurement error yields more than 1200 consistent base compositions. Taking into account the fact that the base compositions of the two strands must be complementary, however, culls the list of putative base compositions to those in which the base composition of “strand 1” is complementary to that of “strand 2.” At a mass measurement error of 20 ppm or less, there are >100 base compositions consistent with the measured masses of each strand but only one combination in which the two strands are complementary.

aMolecular weights were obtained from Figure 3.
bNote that at 20 ppm (or better) the base composition of the amplicon pair is constrained to a single, unique base composition pair (A34G31C29T27 and A27G29C31T34) because the base composition of strand I must be complementary to that of strand 2.
A simple way to visualize the information content in the base composition derived from a single primer pair is illustrated in Figure 4. In this pseudo three-dimensional coordinate system, the number of A's is plotted on the y-axis, the number of C's on the x-axis, and the number of G's on the z-axis. Also plotted (as colored spheres) are the base compositions one would expect to obtain (based on in silico PCR) from the known nucleotide sequences of multiple bacterial strains from various clades. The plot shows that this primer clearly differentiates B. anthracis from other bacterial pathogens shown on this plot such as Streptococcus pneumonia, Staphylococcus aureus, and Streptococcus pyogenes (and thousands of other pathogens not shown). Although B. anthracis is not the only bacterial species that can produce an amplicon from this primer pair having this particular base composition, multiple primer pairs that target different regions of the genome can be applied to determine whether an aggregate base composition signature is consistent with (or inconsistent with) that expected for any given pathogen. Interestingly, even if the aggregate base composition signature has not been observed before, or is inconsistent with all entries in the database, the compilation of these base composition signatures can be used to put the measured signatures into a phylogenetic context. Thus, for pathogens associated with newly emerging infectious diseases (e.g., severe acute respiratory syndrome [SARS]) or bioengineering events that have not been previously characterized, the T.I.G.E.R. method can assign the pathogen to a bacterial species or viral family. At the height of the SARS epidemic, we applied this concept to human coronavirus and demonstrated that the broad-range T5000 coronavirus primers (designed and tested before the emergence of SARS) were able to detect the SARS virus and categorize it as a new member of the coronavirus family. 5, 6
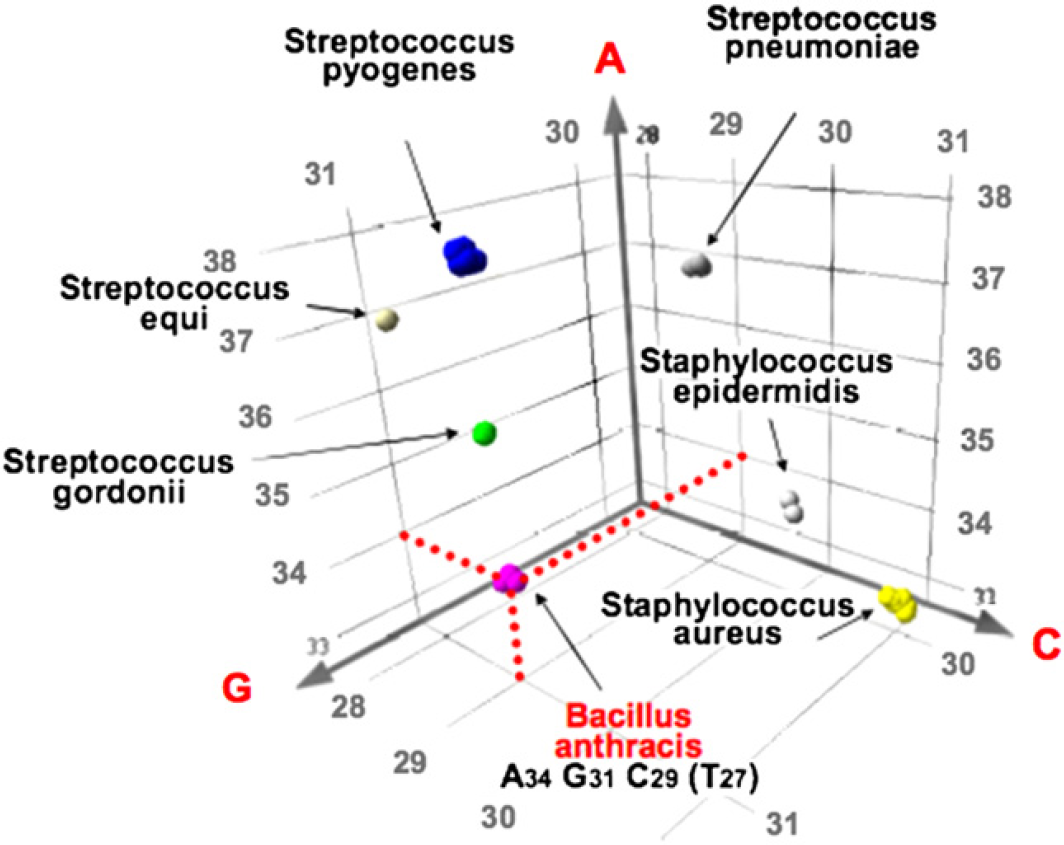
Pseudo three-dimensional representation of the “base composition space” associated with the amplification of a region of rplB from multiple bacterial species. Note that the mass spectrometry–derived base composition A34G31C29T27 (see Fig. 3 for raw mass spectrum) clearly distinguishes B. anthracis from numerous other bacterial species. The collective base compositions derived from multiple broad-range primers amplifying different regions of the genome are used to unambiguously identify pathogens present in a sample.
Sample Preparation
The T5000 process enables the evaluation of a nearly limitless array of microorganisms and viruses. As it is not necessarily clear which organism(s) are in the sample, it is difficult to predict a single best lysis method for each sample. Because the Ibis T5000 system is not dependent on a single sample preparation method, users do have a wide variety of choices for sample preparation ranging from manual methods to automatable methods. Furthermore, it is important to consider the quality of the end products and the efficiencies of the cell lysis and the isolation of the resulting nucleic acid.
During the development of the Ibis T5000 system, we have processed a wide variety of samples. Validated isolation methods for a variety of sample types are listed in Table 2. Sample types include environmental samples (air samples from dry filter units, surface swabs, water samples, etc.), biological samples (bacterial colonies and bacterial or viral cultures), clinical samples (throat swabs, nasal swabs, nasopharyngeal swabs, nasal washes, sputum, blood, and skin swabs), and food samples (meat, dairy, and produce). Efficient lysis is achieved through the use of bead beating and a chemical lysis step involving the use of chaotropic agents. Simple, inexpensive methods such as boiling preparations are also sufficient for use in the Ibis T5000 system. Most of these sample types have been prepared using an automated robotic system with either a viral or a bacterial nucleic acid isolation method based on binding of the nucleic acid to a silica matrix. Recently, magnetic bead–based isolation methods have also been successfully used (Fig. 5).
Sample types and validated nucleic acid isolation methods

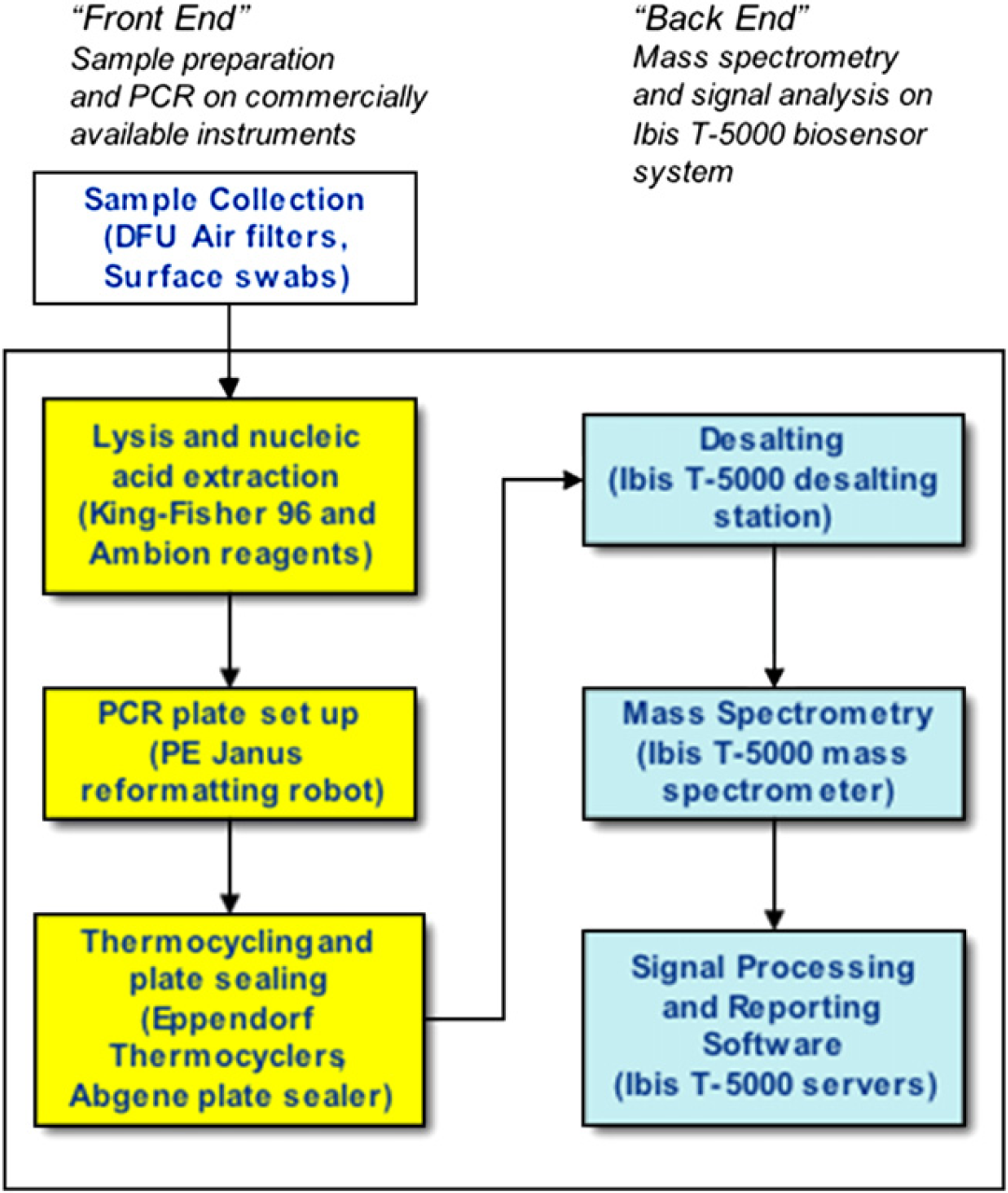
Workflow for sample analysis.
The end-result of the sample isolation process is PCR-ready nucleic acid. The samples often consist of nucleic acid from a mixture of organisms necessitating that the reaction conditions provide unbiased and efficient amplification. Further, as many different sample types and preparation methods are used, the PCRs should be tolerant of interfering components. PCRs or reverse transcription–PCRs are performed using validated, prepackaged T5000 kits. These kits are formulated to achieve the goals of inhibitor tolerance, efficient and unbiased amplification in the presence of potentially mismatched bases, high sensitivity, and robust amplicon production. The kits are also designed to function under universal amplification conditions, allowing the mixing and matching of primer pairs on any given plate-based format.
Each Ibis T5000 kit contains one or more pairs of PCR primers that allow identification of a broad group of pathogens. Ibis T5000 kits containing primers for specific missions and calibration standards have been developed for some of the most important organisms implicated in HAIs, problematic respiratory infections, blood-borne viral infections, and biodefense. In general, multiplexed PCRs allow identification of the pathogens of interest present in a sample in a single mass spectrometry analysis.
After PCR amplification, the 96-well plates are placed on the integrated T5000 platform for automated amplicon purification, electrospray ionization mass spectrometry (ESI-MS) analysis, and signal processing. Below is a brief description of the various hardware modules involved in the ESI-MS analysis of PCR-amplified genomic material.
T5000 Instrumentation Description
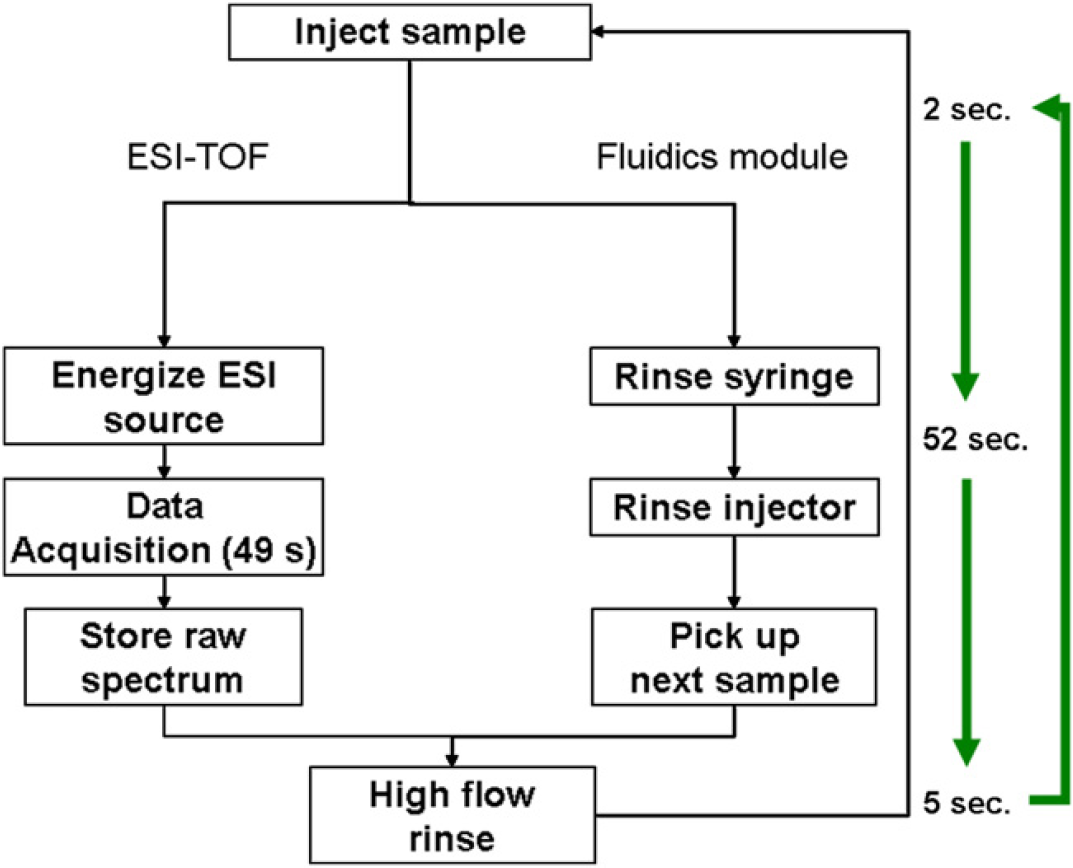
Figure 6 Workflow diagram of the ESI-TOF and fluidics module highlighting the parallel events of the ESI-TOF and the fluidics module used to enhance the overall duty cycle and increase the sample throughput to <1 min/sample.
Software
The software system consists of four components that perform individual functions and interact with instrumentation to provide an automated flow from initial input of samples to analysis results. The four components—robotics system, T-Track sample tracking system, a relational database, and data processor—are represented in Figure 7. A key element of the system is the reliance on the central database; components interact with the database to retrieve and update information throughout the workflow.
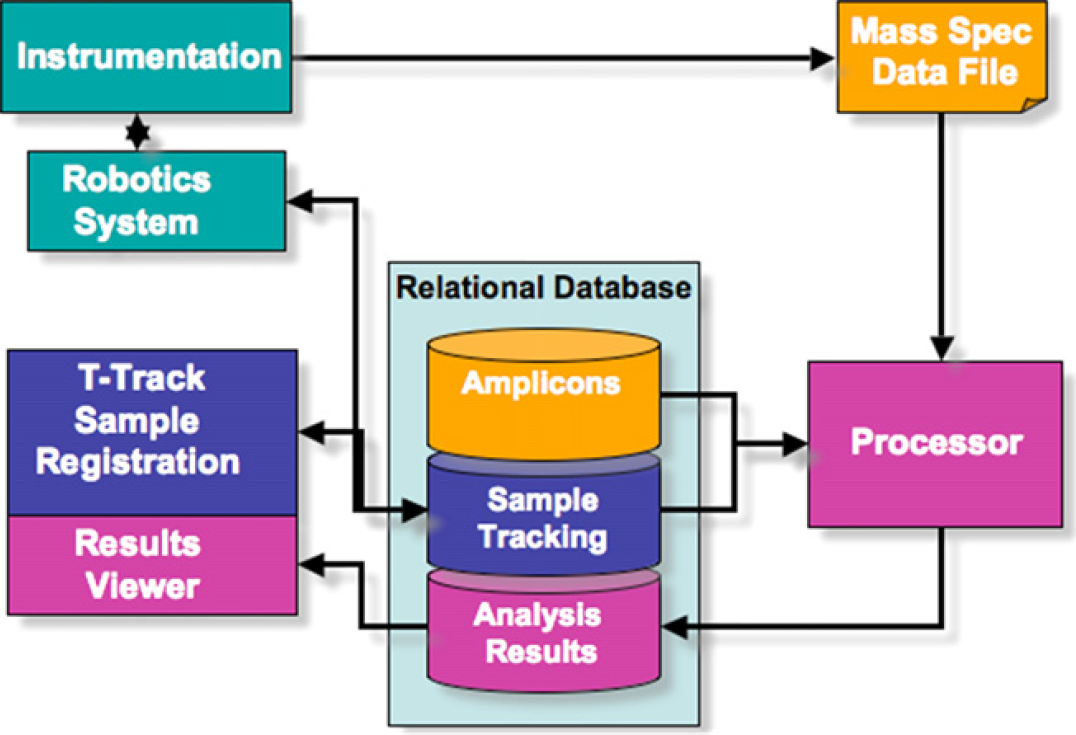
Figure 7 A simplified view of the major components of the Ibis T5000 software system. Components interact to provide a fully automated process from the point samples are placed on the instrument to analysis of results for the user to review.
Upon invocation of the user interface, a main dispatching module is initialized and instantiates a list of devices reflecting the hardware instrumentation. Serial port communication protocols are used for controlling device that comprise the PCR cleanup module, the plate sealer device, and the liquid handling devices. The Liquid Handling and Mass Spec devices rely on an additional slave dispatching module to instantiate and control the mass spectrometer and liquid handlers. Unlike serial communications, low-level software control uses Active-X and DCOM technologies to interface with third-party software to control plate handling and synchronize mass spectrometry acquisition.
After issuing a start from the user interface, the dispatch manager compiles the appropriate method script with specific commands for controlling each device described above. The use of this scripting architecture allows for isolating method development from actual software development and provides flexibility for later optimizations. Additionally, it supports multiple run-modes and incorporation of additional devices using the same controller software.
Along with hardware commands, internal validation commands are embedded within the control method scripts to query information from a database, ensuring plate, sample, and barcode validity. In addition, access to database information enables runtime parameters (e.g., plate map, run-list) to be retrieved directly from the database and minimize user error. File logging and error handling allow for automatic error recovery for certain errors and provide a mechanism for user notification when manual intervention is required.
The workflow in the sample tracking program reflects the kit-based approach to pathogen analysis, in which information on plates that contain PCR primers and reagents, including barcodes, layout, and reagents, is predefined. The user must interact with the system to (1) choose the plan for the experiment, including the assay kit that will be used; (2) select from a list of samples those that will be used; (3) reserve barcodes associated with the kits that will be placed on the instrument; and (4) register plates with associated samples.
The locations of samples as they are distributed from one plate to the next during the process are tracked automatically. The robotics system uses an application programming interface from T-Track to validate and register events during the course of instrument operation. Results from processing are automatically entered into the database and are then available for review, with organism identifications associated with input samples. Results can be located by querying by plate or by the sample identifier, so it is possible to rapidly locate results for a single sample or to evaluate performance of the system on a set of samples.
Masses of all amplicons in a sample are determined from raw spectral data. Potential base compositions are assigned to all masses within acceptable error levels. As complementary strands are present, this restriction is used to limit the possible choices of base compositions consistent with molecular weight (Table 1). These base compositions are then used as hypotheses, from which a spectral representation is modeled. At this point, organisms consistent with the primer pair used in PCR amplification are identified from the amplicon database. A joint least-square algorithm is used to correlate potential organism identifications across multiple primers, using a triangulation method. This computation process improves the confidence of correct organism classification and reduces the possibility of false positives.
Applications
The Ibis T5000 instrumentation and associated kits have a wide variety of uses. Basically, the instrumentation can be used in any application where detection of a biological agent is needed. In addition, human forensic identification using either mitochondrial or chromosomal markers can be examined, where mass spectrometry is used in place of sequencing or gel electrophoresis, respectively. 8 Table 3 summarizes applications for which the Ibis T5000 is currently being used or developed, and representative examples of its use are described briefly below.
Applications of the Ibis T5000 instrument

HAI, hospital acquired infection; SARS, severe acute respiratory syndrome; MRSA, methicillin resistant Staphylococcus aureus; VRE, vancomycin resistant enterococcus; HVAC, heating, ventilation, and air conditioning; CDC, Centers for Disease Control and Prevention; FBI, Federal Bureau of Investigation; DHS, Department of Homeland Security; FDA, Food and Drug Administration; USDA, United States Department of Agriculture
Epidemiological Surveillance
We studied sick military recruits at the Marine Corps Recruiting Depot (MCRD) in San Diego during the most severe outbreak of streptococcal disease to have occurred in the United States since 1968. During the outbreak, over 160 recruits were hospitalized and one death had occurred. 9 We analyzed throat swabs from sickened recruits to address two questions: First, what pathogens and copathogens were responsible for this severe epidemic and second, could our methodology be used to follow the spread of the epidemic at MCRD and in other military facilities. Analysis of respiratory samples revealed high concentrations of several pathogenic respiratory bacteria, including Haemophilus influenzae, Neisseria meningitidis, and S. pyogenes. 6 When S. pyogenes 10 was identified in samples from the epidemic site, the identical genotype was found in almost all recruits, consistent with a model of clonal expansion of a severe pathogen (Fig. 8). We also examined samples from five other military bases, and these locations showed a pattern significantly different from the MCRD epidemic, suggesting that the epidemic strain was not spreading to other military facilities. The Ibis T5000 Biosensor system provided analyses with sufficient speed and throughput to be useful in tracking of an ongoing epidemic and provided information fundamental to understanding the polymicrobial nature of explosive epidemics of respiratory disease.
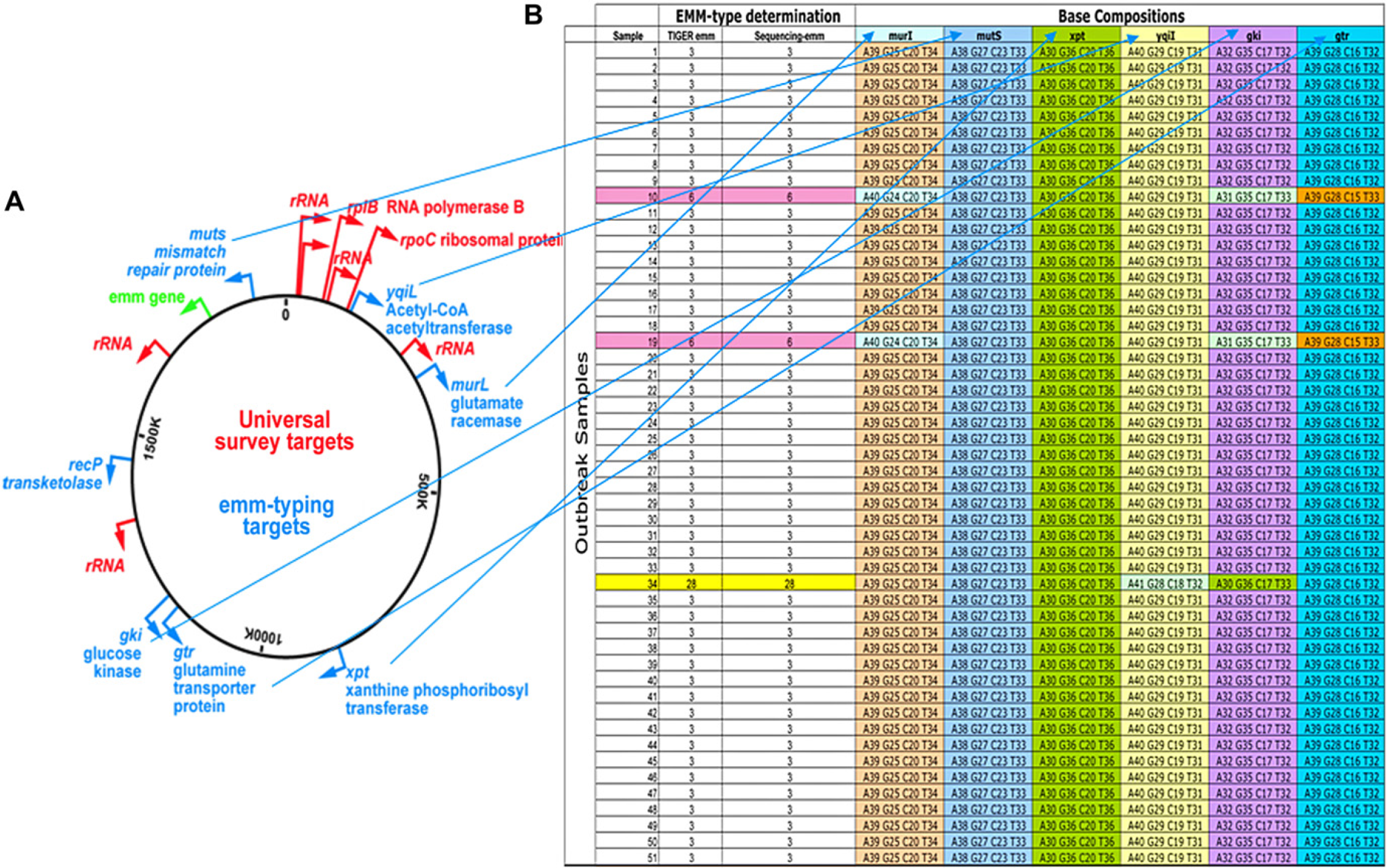
Identification and bacterial strain genotyping using the Ibis T5000 primers and base composition signatures. (A) Using primers that target genes that are broadly conserved across all bacteria (red), bacteria were first identified at the species level; subsequently, genotyping primers (blue) were used to target variable regions of household genes that are highly conserved among a given species (S. pyogenes samples from a pneumonia outbreak in this example). The resulting base compositions tabulated in (B) provided a base composition “barcode” that yielded a specific strain-type to allow tracking of the epidemic.
HAI Control
HAIs, the fourth leading cause of disease in the United States, are a major public health problem accounting for approximately 2 million infections, 90,000 unnecessary deaths, and $4.5 billion in excess health care costs in the United States each year. Although the numbers of patients treated in hospitals and the average length of stay decreased during recent decades, hospital-acquired infections have increased. Further, the consequences of hospital-acquired infections are more severe than they were a decade ago due to emerging antibiotic-resistant microbes, especially S. aureus, Enterococcus, Acinetobacter, Pseudomonas, Klebsiella, and Acinetobacter species.
Aggressive control measures can slow and usually halt the spread of difficult-to-contain infectious agents. Typically, control of HAIs relies on identifying the infectious agent and its genotypic properties, obtaining information on the numbers of potentially exposed patients and their environment, and then applying appropriate infection control measures. Shortages of tools for quickly and fully identifying and characterizing microbes hamper deployment of infection control measures. For instance, screening microbes with culture methods requires 24–72 h, is labor intensive, is technique-dependent, and has limited sample throughput. Most critically, culture techniques do not provide the details that those in charge need to react appropriately. Typically, additional rounds of testing on isolated colonies or enriched samples are required to determine drug-resistance or genotype information, further delaying corrective steps while adding to overall costs. Meanwhile, patients may be unnecessarily isolated, or contact control measures may be delayed, sometimes enabling infections to spread throughout a hospital.
Technologies for improving infection control measures in hospitals should incorporate several properties, including speed, ease of use, high reliability, and an ability to detail a microorganism's molecular profile. A primary requirement is that the identity of the microbes, along with their important genotypic features, be provided rapidly. This requirement rules out using culture steps and, instead, requires direct analyses. To analyze such large numbers of samples, labs must be equipped with high-throughput automated systems that provide all relevant information on microorganisms being analyzed, including species, subspecies, presence of antibiotic resistance genes, virulence factors, and/or toxin-encoding genes.
An example of the power of the Ibis T5000 in an infection control setting was demonstrated by examining an Acinetobacter outbreak in collaboration with Department of Defense researchers at Walter Reed Army Medical Center. Acinetobacter is a ubiquitous natural soil and water bacterium historically associated with infections in wounds obtained from combat injuries. It has become an important nosocomial pathogen worldwide. We characterized 217 isolates collected from soldiers and civilians from six military field hospitals in Iraq and Kuwait, two large tertiary care hospitals in Landstuhl (Germany) and Washington, D.C. (USA), and a hospital ship in the Persian Gulf. 11 We used the Ibis T5000 method to identify the molecular genotype of each isolate and to determine patterns of clonal expansion. We also compared these isolates with 23 previously characterized reference strains from outbreaks in European hospitals and 19 reference strains from culture collections. The Ibis T5000 analysis revealed 76 unique strain genotypes. The vast majority (190/217) of the clinical samples associated with the conflict in Iraq were Acinetobacter baumannii strains with genotypes very similar or identical to previously characterized multidrug-resistant (MDR) A. baumannii isolates from European hospitals. All of the major clusters (multiple isolates with identical genotypes) were members of this group. A smaller fraction (27/217) had previously unencountered genotypes that were evolutionarily far removed from the European hospital strains. The small number of previously unencountered isolates may represent de novo infections of wounds from the soil. The primary source of these health care–propagated outbreaks is likely to be the introduction of MDR A. baumannii strains into the military hospitals from military casualty hospitals in Iraq (Fig. 9). This study demonstrates the value of this novel technique in evaluating complex outbreaks at the molecular level and the difficulty involved in maintaining infection control procedures in a combat environment.
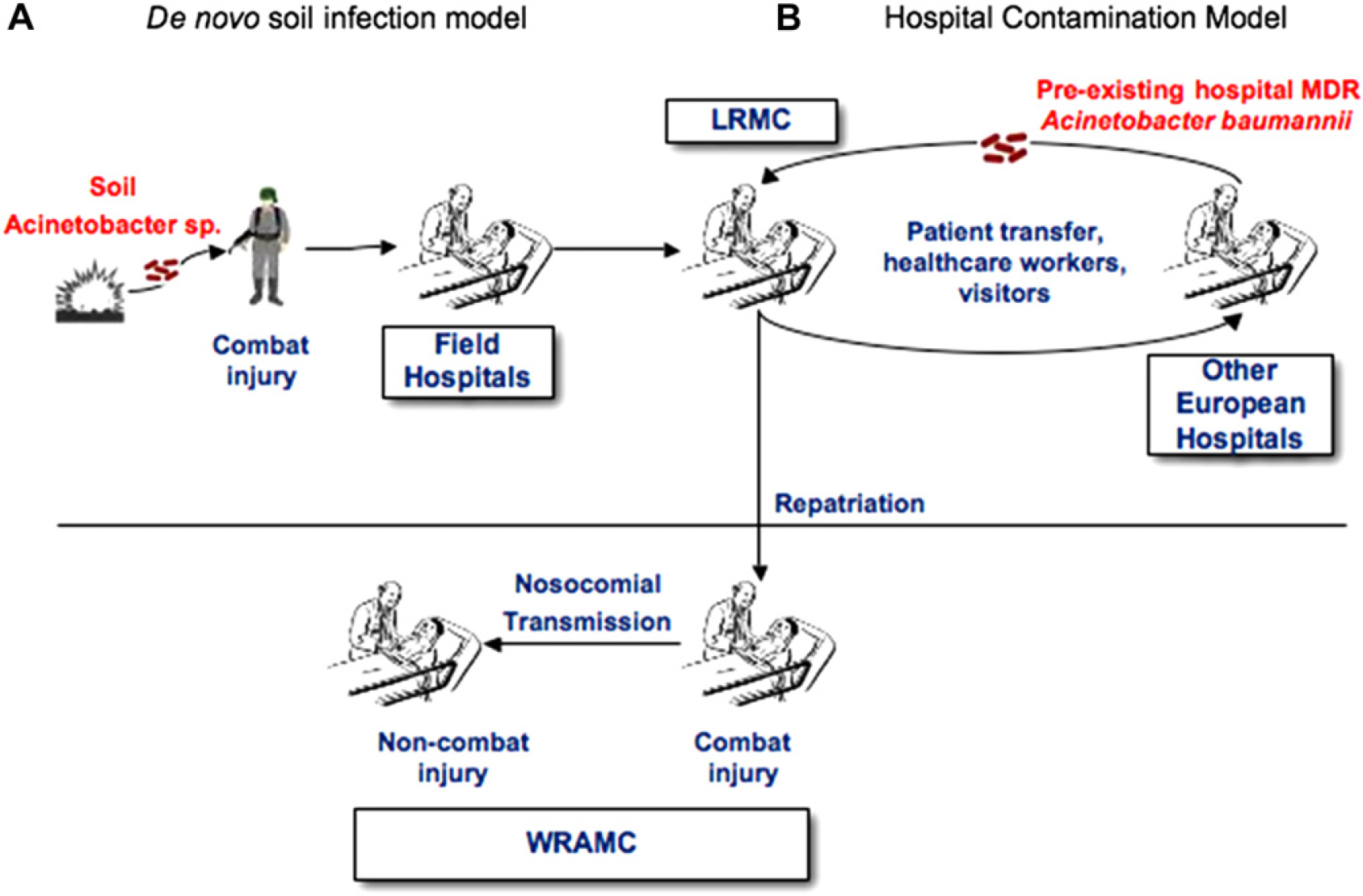
Alternative models for the source of the infection. (A) Acinetobacter species present in soil infects soldiers wounded in combat as a de novo infection. (B) Preexisting MDR A. baumannii from European hospitals enter the US military hospital system. The Ibis T5000 genotyping data strongly support the latter model.
Conclusions
We have demonstrated that the T.I.G.E.R. strategy can be applied in a highly integrated and automated system. The Ibis T5000 is capable of detecting and identifying a wide variety of microbes including bacteria, viruses, fungi, and protozoa. Due to the high degree of automation and software control, it is intended to be operated by a properly trained laboratory technician with no formal training in mass spectrometry, signal processing, or molecular biology. Broad-range PCRs are capable of producing products from groups of organisms, rather than single species. The ability of the mass spectrometer to rapidly and accurately derive base compositions from PCR amplicons provides high information content from each reaction and obviates the need to anticipate the exact nature of an amplicon with sequence-specific probes. The T5000 is capable of analyzing complex PCR products at a rate of approximately one well per minute; thus, it is possible to examine large numbers of samples, making it practical for large-scale analysis of clinical specimens or for environmental surveillance.
Acknowledgment
This work was funded in part by DARPA under contract MDA972-00-C-0053 and by the CDC under grant #1R01CI000099-01.