
Abstract
An automated platform for low-volume, highthroughput protein crystallization by the sitting drop vapor diffusion has been developed. This platform consists of a novel Society of Biomolecular Screening standard microplate technology, standard automation liquid handling equipment, a software to design customizable, random crystallization buffers, an in-house designed scanner, a crystal recognition software, and Laboratory Information Management Systems. These technologies are designed to eliminate obstacles to the production of protein crystals and to address a number of critical issues faced by crystallography laboratories, namely precision and accuracy, ease of integration and use, speed, and cost.
Keywords
Introduction
The three-dimensional structures of proteins provide insight into their physiological functions, facilitating the design of more efficient drugs and therapeutic agents. The two major platforms currently used to explore the structure of proteins are X-ray crystallography and NMR spectroscopy. Advances in high-throughput protein crystallography have outpaced developments in NMR technologies, because the development of automation and other high-throughput technologies toward achieving maximal efficiency in this field has been more rapid. Furthermore, NMR studies generally do not provide structural information for proteins larger than 25 kDa.
Many different institutions are actively engaged in the development of robotic technologies for automation of all the stages involved in protein crystallography (Fig. 1). These stages range from the preparation of the proteins (from cloning, to expression, to purification), to crystallization (screening and crystal optimization), to X-ray diffraction screening, and finally, to structure determination (bioinformatics and deposition in structure databases).
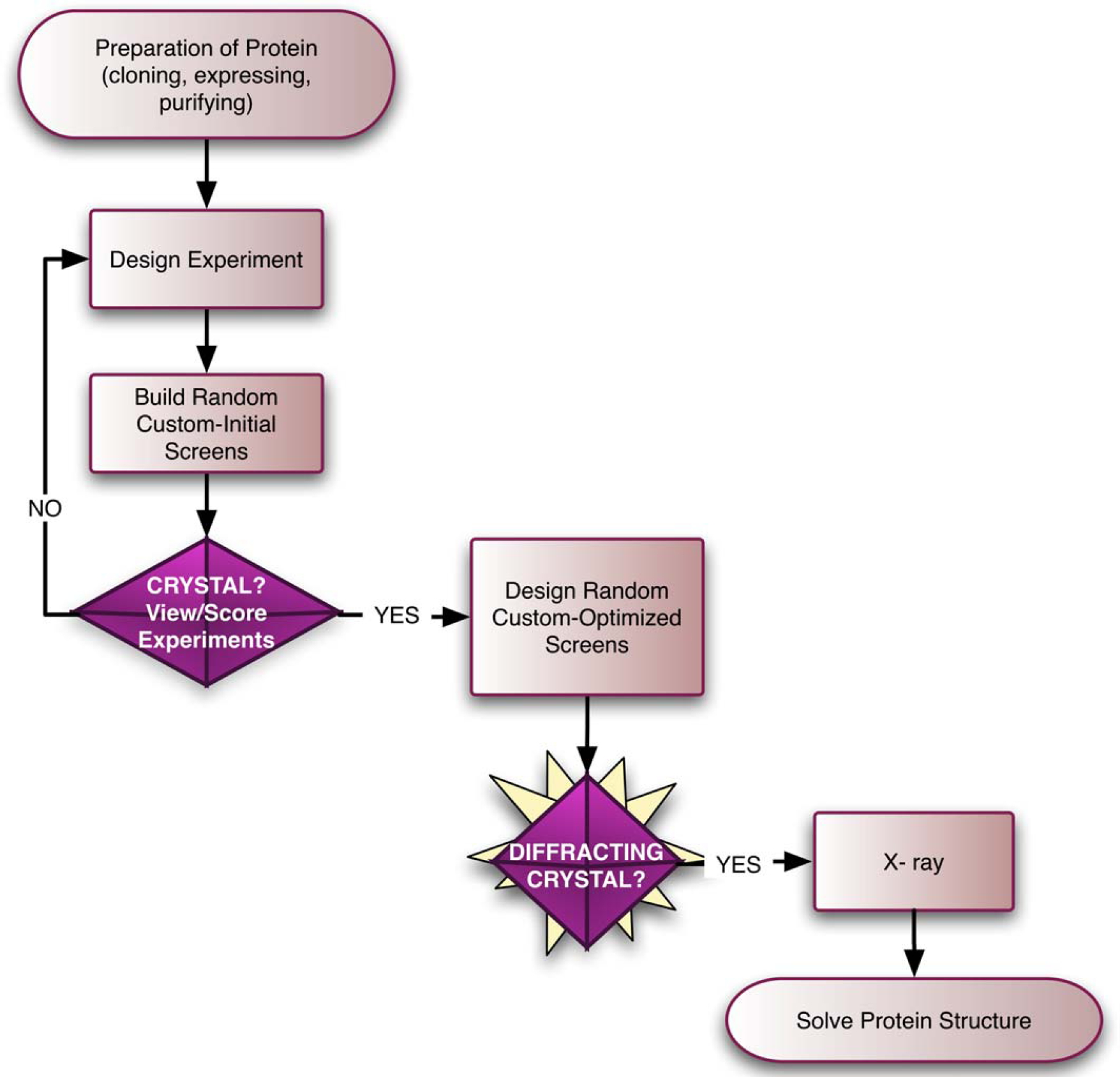
Structural genomics process. The gene of interest is cloned, expressed, and the protein is purified. Initial crystallization screening experiments are next set up by sitting drop, hanging drop, or microbatch. Based on initial screening results, diffracting crystals are produced using custom-optimized screens. Crystals are harvested, frozen, and their diffraction properties analyzed by X-ray. The three-dimensional structure of the protein is determined based on the diffraction data collected.
Proteins are generally scarce in natural sources and costly to isolate and purify in quantities sufficient for crystallography studies. Months of effort are often needed to purify a few micrograms of a desired protein. As a result, automated crystallography platforms have emerged aimed at miniaturization to limit the use of proteins down to the nanogram range. These approaches present multiple challenges. Given the complexity (for instance the solubility and viscosity) of proteins and the screening (crystallization) solutions (mixtures of acid, bases, detergents, salts, alcohols, etc.) utilized, the task of creating workstations to dispense solutions in the submicroliter range with high precision and accuracy is non-trivial but none the less vital.
Another challenge is to design a crystallization method amenable to automation, which would provide diffracting quality crystals, using submicroliter volumes. The most popular protein crystallization methods are vapor diffusion by sitting drop, hanging drop, and microbatch. 1 Sitting drop technology is the most automatable and thus, more commonly used by crystallographers (Fig. 2). A typical sitting drop platform (plate) used with automation conforms to the Society of Biomolecular Screening (SBS) standard dimensions and contains 96 buffer reservoirs (the large wells), each with a corresponding protein well (the small well). When the protein and the buffers are dispensed and the plate is sealed, the starting concentration of the protein drop in the protein well is half the concentration of the crystallization screens. The diffusion of solvent in the vapor phase from the protein well to the buffer reservoir gradually increases the protein concentration and drives protein precipitation, hopefully resulting in crystallization. 1,2
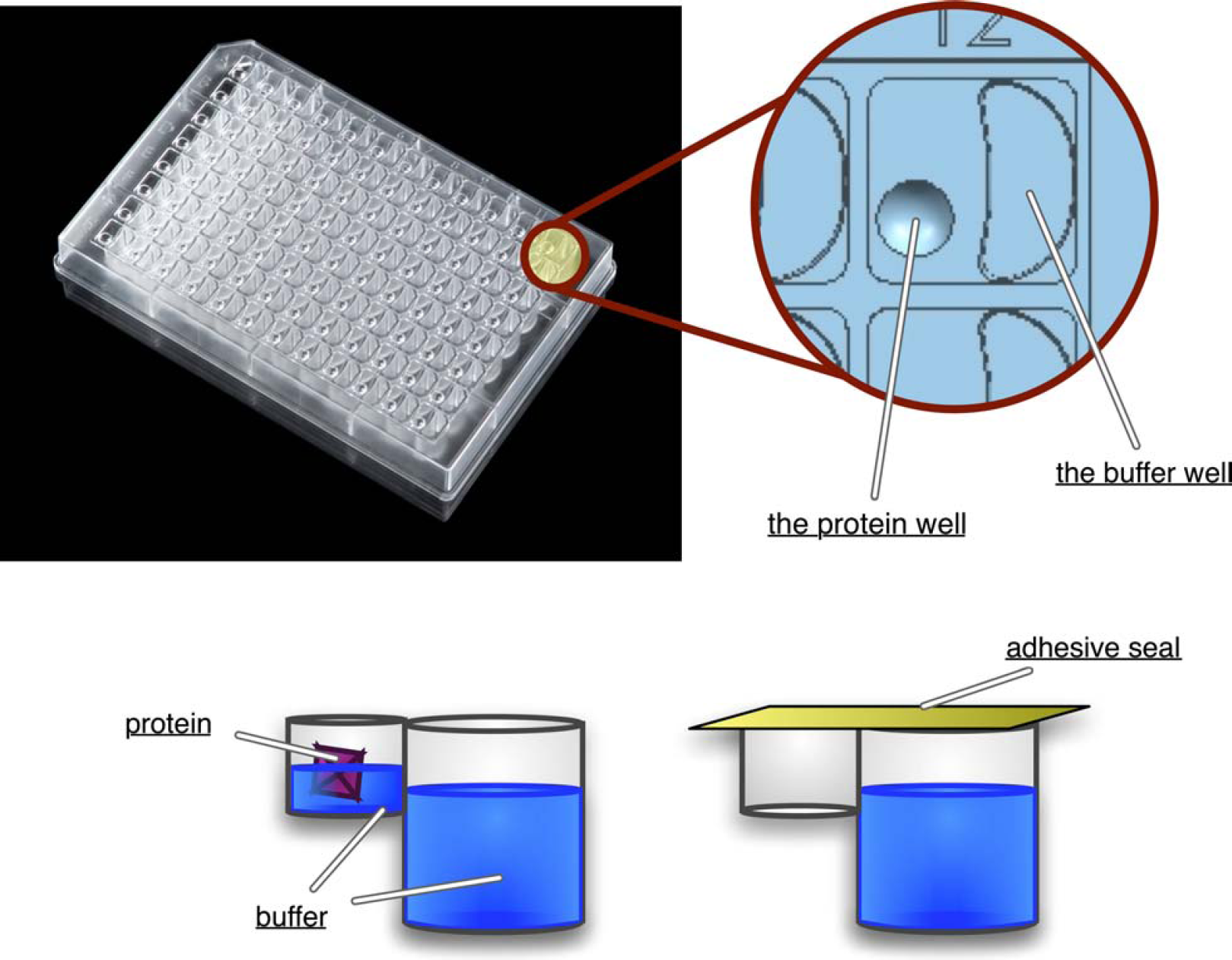
Crystallization in a 96-well sitting drop crystallization plate. Crystallization buffers (screens) are added to the reservoir as well as to the protein well. Protein is added to the protein well and the plate is sealed. The concentration in the reservoir is twice the concentration in the protein well. Evaporation occurs from the protein well to the reservoir. The diffusion of solvent in the vapor phase from the protein well to the reservoir gradually increases the protein concentration and thus drives protein crystallization.
The standard workflow of protein crystallography involves the preparation of crystallization buffers. The objective is to use factors such as salt concentration, pH, and temperature in combination to arrive at an ideal condition that induces the formation of crystals. Potentially useful combinations of conditions number in the 10s to 100s of millions but are tedious, labor-intensive, and time consuming to produce as screens, and only a few of these combinations will result in successful crystallization for any given protein. This emphasizes the importance of creating a limited number of efficient crystallization conditions because the availability of stock protein, cost of screens, and storage issues prevent the generation of a large number of screening conditions.
Once the crystallization screens are designed and the sitting drop experiments are set up, the next task involves the detection of micron size crystals. Observing and scoring thousands of crystallization experiments quickly becomes the most labor-intensive part of the workload. The solution is to automate the imaging as well as the scoring steps. The challenge lies in accurately detecting crystals and determining the difference between crystal and precipitate, without making any false-negative/positive assessments.
When crystals are detected in the screening process, the initial conditions are stored in databases and optimized to generate diffracting quality crystals. These crystals are harvested and frozen for X-ray analyses. Their diffraction pattern data are analyzed by sophisticated algorithms to solve their three-dimensional structures. The information generated from each of the multiple steps is stored in Laboratory Information Management Systems (LIMS) or databases for further improvement and tracking.
It is estimated that the costs of collecting structural data for a given protein are in the quarter of a million dollar range. In addition to the economical constraints, the speed of attaining the structure of a protein (ranging from months to years) imposes another major obstacle. Therefore, to achieve maximal overall efficiency to implement successful high-throughput crystallography, there is a need to design cost-effective, optimized, faster, and simpler platforms composed of integratable workstations, robots, and databases. The purpose of this study was to generate an automated platform to set up low-volume, high-throughput protein crystallization by the sitting drop vapor diffusion technology. Components of this platform individually and in unison are chosen or designed to increase precision, accuracy, and speed, while optimizing the ease of use, integration, and cost efficiencies.
Materials and Methods
The Axygem™ plates (CP-Axygem-96), UltraClear™ pressure-sensitive adhesive sealing film (UC-500), Semi-Automated Protein Crystallography Plate and Microplate Sealer (MS-100), CRYStool™ software (AB-CRYS-TOOL), AxyScanner™, and Crystal Recognition Software (CRS™) (AB-CRS) were provided by Axygen Inc., Union City, CA. The Hydra®-Plus-One system (composed of the Hydra®-PP dispenser equipped with 96, 100 μL syringes with Teflon®-coated stainless steel needles and the Nanofill® single-channel, noncontact microsolenoid dispenser) was purchased from Robbins, Sunnyvale, CA. The eight-channel MultiPROBE® II HT system was purchased from Perkin Elmer Life Sciences. The Motoman Arm integrated with a plate hotel, providing 1000 plate storage capacity, was purchased from Willamette Valley Company, Oregon.
Protein Crystallization
Crystals of Thaumatin protein were obtained by the sitting drop vapor diffusion method. The experiments were set up at 22 °C using 1.5 μL of protein (37.5 mg/mL) and 1.5 μL of crystallization solution (mother liquor) suspended over 200 μL of crystallization buffer. Crystals (∼0.3 mm) grew overnight with 0.1 M HEPES pH 7.5 and 0.8 M sodium potassium tartrate as the mother liquor. Crystals continued to grow for several days reaching ∼0.7 mm in size.
CRYStool

A high-throughput platform for low-volume protein crystallization by the vapor diffusion sitting drop technique. The
Preparing the Dispensing Systems: The MultiPROBE II and the Hydra-Plus-One Systems
The MultiPROBE II microdispenser system was used to produce the random screens generated by CRYStool (Fig. 3). This system can operate with disposable tips eliminating the need for cleaning procedures, reducing the possibility of cross-contamination and increasing the speed. The Hydra-Plus-One system 5 (Fig. 3) was used to set up the 96-well crystallization microplates. The Hydra-PP part of the Hydra-Plus-One system, equipped with 96 fixed needles, was used to simultaneously dispense the 96 different crystallization buffers first into the D-shaped buffer reservoirs of the crystallization plate and then into the protein well. The NanoFill single-channel dispenser part of the system was used to dispense the protein into each of the 96 protein wells. To prevent carry-over and cross-contamination, stringent cleaning procedures were followed for both the syringes and the microsolenoid dispenser (protocol published elsewhere). 6,7
The dispensing precision of the Hydra system, the single-channel microdispenser, and the MultiPROBE was determined (for volumes of 100 nL–10 μL for the Hydra and the microdispenser, and 2-10 μL for the MultiPROBE) by the coefficient of variance (CV = standard deviation/mean) (detailed protocol reported in references 8 and 9). In short, different volumes of a 10 μg/mL fluorescein solution were dispensed into each well of a 96-well plate and the volume of each well was adjusted to 100 μL with 0.1 M Tris buffer. Each plate was read in a Tecan fluorescence plate reader, and the CVs were determined for each of the volumes dispensed. Uniformity and consistency for the dispensed volumes were evident with CVs of less than 10%.
Sealing of the Crystallography Plates
The Protein Crystallography Plate and Microplate Sealer was used for the adhesive sealing of crystallography sitting drop plates by cold pressure (Fig. 3). This system is an electromechanical sealing device containing a pressure block (which could be used at room temperature or at an adjustable temperature) and cycle duration control panels. The pressure block comes into contact with the adhesive seal (placed on top of the crystallography plate) for a period of a few seconds (adjustable) and evenly seals the entire plate.
Results and Discussion
An automated platform to set up low-volume, high-throughput protein crystallization by the sitting drop vapor diffusion technology has been implemented in this study. Components of this platform are composed of a screen-designing software tool called CRYStool, a micro and a nanoliter dispenser to set up screens and the crystallography reactions called the MultiPROBE and the Hydra-Plus-One systems, respectively, a plate sealer called the Protein Crystallography Plate and Microplate Sealer, a robotic arm with plate hotels called the Motoman Arm, an imaging system equipped with CRS called the AxyScanner, and two LIMS systems for data storage, analysis, and improvement purposes. The efficiency of this platform in overcoming the current limitations in crystallography was analyzed by parameters such as cost, speed, sample consumption, precision, and ease of integration.
Designing Initial Screens Using CRYStool
The first step in protein crystallography, considered to be the rate-limiting step in structural studies of proteins, 10 involves the preparation of crystallization screens. A number of factors, such as salt concentration, pH, and temperature, can be used in combination to create millions of individual screens. However, only a few of these conditions are capable of inducing crystal formation. The availability of stock protein, cost of screens, time, and storage issues emphasize the importance of generating a limited number of efficient conditions.
CRYStool is an automated proprietary algorithm enabling the creation of highly efficient, customizable, random crystallization screens that efficiently sample the multidimensional space of crystallization conditions. This software was developed at Lawrence Livermore National Laboratory (LLNL), a participant in one of the nine government-founded Protein Structure Initiative centers. CRYStool was initially developed to determine which of the screening protocols commonly used for crystallization—screening by random combination (inspired by Charles Carter's incomplete factorial and Jankarick and Kim's sparse matrix), 11 footprint screening (inspired by Erico Stura's footprint screen), 12 and grid screening (inspired by Alex McPherson's grid screening) 1,13 —was the most efficient. Screening by random sampling was the most successful route in that the fewest number of screens were needed to crystallize proteins. 4 In this study, it was estimated that a crystallizable protein has a greater than 99% chance of crystallizing using 288 randomly chosen conditions generated by CRYStool. If crystallization is not achieved after 288 trials, it is reasonable to consider altering the protein rather than continuing to screen to induce crystallization.
In this study, the CRYStool software was easily integrated into a microliter dispensing system called the MultiPROBE II (PerkinElmer) (http://www.perkinelmer.com) to produce the initial 288 random screens to crystallize a protein called Thaumatin. This system has four to eight probes working with disposable tips and can be used to set up CRYStool screens in a 96-deep-well polypropylene plate. It takes approximately 2 h and 15 min to set up a full CRYStool random screen (288 screens per 3 plates) from 90 premixed stock solutions.
CRYStool software can also be integrated into other dispensing equipments such as the Evo system from Tecan (a microliter dispenser) (http://www.tecan.com), Screenmaker 96 + 8™ Xtal from Innovadyne (a nanoliter dispenser) (http://www.innovadyne.com), or the Matrix Maker from deCODE (a microliter dispenser) (http://www.decode.com) to produce crystallization screens. These dispensing instruments range in price from approximately $65K (Multi-PROBE) to $200K (Screenmaker 96 + 8™ Xtal) depending on the speed of dispense (from 10 to 45 min per 96-deep-well plate) as well as the volume (nano or microliter range) and precision (CVs of less than 10%) of dispense.
Setting up Crystallization Plates
An SBS-standard, 96-well sitting drop plate, called the Axygem plate, was designed to set up low-volume, high-throughput protein crystallization. This plate is made of optically clear polystyrene to facilitate precise crystal visualization. It is static free (nonchemically treated) and individually wrapped in antistatic film. It has 96 buffer wells (maximum volume of 250 μL) and one adjacent protein well (maximum volume of 4.3 μL). The buffer well has a novel D-shaped geometry to decrease the “shadowing effect” interfering with proper optics, and thus optimizing crystal visualization. The elevated round protein well has a lens-shaped design to facilitate crystal retrieval and manipulation and to provide superior optics. In addition, the round design of the protein well as well as the static-free nature of the plate prevents the separation of the protein and the buffer drops upon dispense, and thus the need for lengthy centrifugation procedures. Furthermore, the plate is designed to include a cavity near each protein well to allow the manipulation of crystals (such as for cryosoak and rinsing purposes), eliminating the need to transfer crystals to yet another protein well (Fig. 2).
This new plate is compatible with automated robotic equipments (arms, dispensers, sealers, scanners, etc.) and can be used to set up crystallization reactions in the nano (as low as 100 nL) to microliter (as high as 4 μL) range. The Hydra-Plus-One (Fig. 3) system (approximately $75K), a highly precise nanodispenser (dispensing precision of less than 10% for volumes equal to and greater than 100 nL), was used to dispense the crystallization buffers (from the 96-deep-well plates) and the protein (from a microtube) into the crystallography plate. Total set up time for one (96-well) plate using the Hydra-Plus-One was 4 min including a 2-min wash procedure. Crystals of Thaumatin protein were obtained in this plate using 1.5 μL of protein (37.5 mg/mL) and 1.5 μL of crystallization solution (mother liquor) suspended over 200 μL of crystallization buffer (Fig. 4) (all experiments were performed in triplicates).

Crystallization. Crystals of Thaumatin protein were obtained by sitting drop vapor diffusion method using the Axygem plate. The experiments were set up at 22 °C using 1.5 μL of protein (37.5 mg/mL) and 1.5 μL of crystallization solution (mother liquor) suspended over 200 μL of crystallization buffer. Crystals (∼0.3 mm) grew overnight with 0.1 M HEPES pH 7.5 and 0.8 M sodium potassium tartrate as the mother liquor. Experiments were performed in triplicates.
Plates were also set up using other commercially available dispensers ranging in price from $75 to $200 based on performance criteria of dispensing volume (nano to microliter range), precision, and speed of dispense. MultiPROBE from PerkinElmer, Evo system from Tecan, Screenmaker 96 + 8™ Xtal from Innovadyne, and the Phoenix Liquid Handling System from Art Robbins Instruments (http://www.artrobbinsinstruments.com/) all performed well. The Hydra-Plus-One and the Phoenix systems were the most economically efficient (in addition to low cost, they work with nondisposable probes, eliminating the cost associated with consumables) dispensing technologies enabling fast and high-precision nanoliter dispensing. In addition, protein waste or dead-volume, associated with aspirating protein solutions from a microtube and dispensing it into each plate by the microsolenoid dispenser, is almost eliminated (approximately 1% per experiment) for the Hydra-Plus-One system.
The plates were sealed with an optically adhesive seal and an automated sealer. Sealers also vary in price and speed, being semiautomated (valued at less than $10K) to fully automated (starting price of $20K). In this experiment, a semi-automated sealer (approximately $8K) from Axygen called the Protein Crystallography Plate and Microplate Sealer was used to seal a plate in less than 3 s. Plates were handled and stored in hotels using the Motoman Arm. Thaumatin crystals grew overnight with 0.1 M HEPES pH 7.5 and 0.8 M sodium potassium tartrate as the mother liquor (initial screen) designed by CRYStool (Fig. 4).
Imaging the Crystallization Plates
An in-house imaging system called the AxyScanner was developed to scan the crystallization plates. This robotic CCD microscope was designed to provide precise positioning, motorized zoom and focus, and a reconfigurable light system enabling both bright and dark fields. The imager is equipped with a 4 Mpixel air-cooled CCD camera to maximize signal-to-noise ratio and 3×-21× motorized optical zoom. A pattern recognition software called CRS, developed at LLNL, was integrated into the AxyScanner. The CRS algorithm detects the difference between crystal and precipitate with an accuracy of higher than 97% compared to a trained human observer (Fig. 5). The main advantage of the CRS software is its ease of integration into existing scanners. The cost associated with the AxyScanner is approximately $65K making it economically and performancewise a very efficient imaging system (most existing imaging systems are priced in the $100K range and only a few include a pattern recognition software).
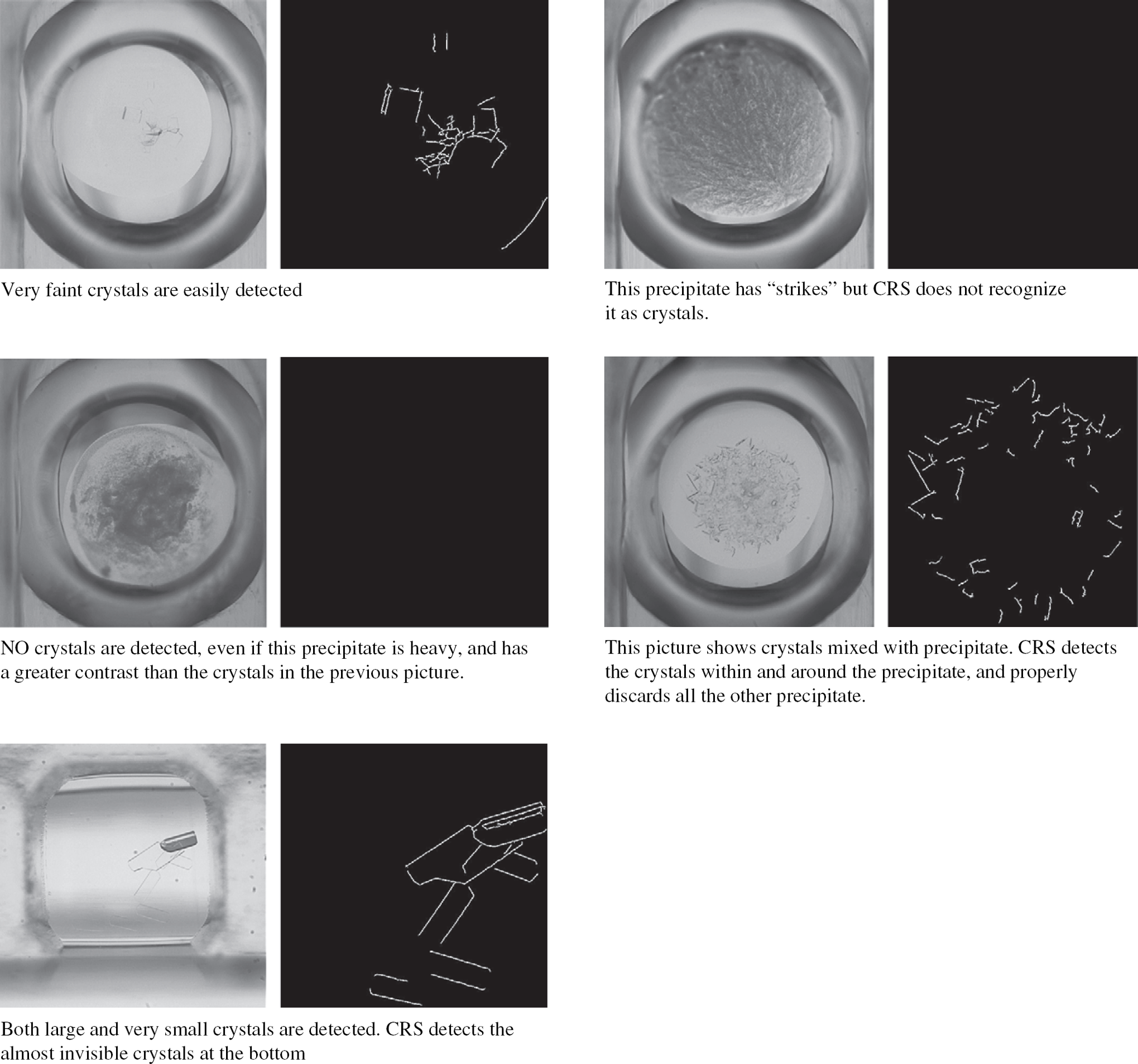
The CRS. The CRS detects very faint crystals and the difference between crystal and precipitate.
Plates stored in hotels are grabbed by the Motoman Arm at fixed intervals and placed on the AxyScanner stage. The plates get scanned and the images get processed with CRS. Once the plate is scanned, the Motoman Arm returns the plate to its original hotel storage site. The data obtained from the scans as well as all the screen information generated by the CRYStool software are stored in two LIMS systems for tracking and improvement. Once enough information is generated for different proteins, the data collected in the databases can be used to facilitate future crystallizations of other valuable proteins.
The Throughput
Using the designed platform, a crystallographer can use the CRYStool software to design screens by choosing a few parameters (based on the available information on a particular protein) and generate a full set of screens using the Multi-PROBE dispenser in a period of 2 h and 30 min. A Hydra-Plus-One system can be used to set up one sitting drop crystallization experiment in 4 min. This translates into approximately 15 plates per hour. The plates can be sealed in a few seconds and transferred to hotels and stored for up to 6 months. The plates can be transferred with the robotic arm onto the scanner stage six to eight times over the active life-span of a plate. Each plate is scanned in a few minutes and the image scored by CRS in a few seconds. As many as 100 plates per day can be generated and processed by this procedure.
Conclusion
In this study, an automated high-throughput platform, consisting of software, databases, dispensing, scanning, and robotic arm equipments and a novel sitting drop plate technology, allowed the generation of large-scale, low-volume crystallization experiments. The components of this platform were designed or chosen based on precision performance issues, cost and time efficiencies, and ease of integration.
CRYStool and CRS software tools were designed to be easily integratable into any existing dispensing and scanning technologies, respectively. Numerous crystallography laboratories have spent thousands of dollars in purchasing such technologies and to improve their existing procedures could use these software tools rather than spend more money in purchasing new equipments. Whereas CRYStool increases the chances of crystallizing a protein, CRS increases the chances of automatically and accurately detecting micron size crystals in a fraction of the time required to observe each protein well under the microscope or view each image on a computer screen.
The dispensing robots were chosen based on the volume (nanoliter), speed, and precision of dispensing as well as the cost. The scanner system was designed to improve crystal detection while lowering the cost and time of imaging thousands of crystallization experiments. The new plate technology was developed to increase throughput (because it can be handled by robots), improve optics, and eliminate static-related issues interfering with successful crystallization of proteins. The LIMS systems were generated to ease the tracking of the large amount of data produced and to consequently improve future crystallization of other proteins.