
Abstract
Theoretical studies focusing on the nature of landscapes that correlate molecular sequences to molecular function have mainly been carried out in silico due to the vast amounts of data that are needed. Automated in vitro selection is capable of producing significant amounts of data in a short time, making theoretical modeling with real experimental data attainable. A Biomek 2000 Laboratory Automation Workstation has been outfitted to carry out multiple in vitro nucleic acid selections in parallel, yielding substantial amounts of data for theoretical studies. A random sequence population of nucleic acids is initially generated by a combination of chemical synthesis and enzymatic amplification. On the workstation, this population is parsed for its ability to bind a protein, lysozyme. After each round of selection, the selected nucleic acid binding species (also known as aptamers) are amplified by a combination of reverse transcription polymerase chain reaction (PCR) and in vitro transcription. All eight pools that have undergone selection have yielded different sequences.
Introduction
The nature of the landscape that correlates molecular sequence to molecular function has been the focus of a number of evolutionary theorists. These theoretical studies have primarily been modeled in silico due to the vast amounts of data needed that were deemed unattainable through the traditional wet-lab approach. Theoretical biologists have speculated for many years on the nature of fitness landscapes, such as how sequence space maps to any of a number of different phenotypes (including folding, catalysis, and substrate specificity). For example, Schuster and his coworkers have used in silico models to exhaustively examine how nucleic acid sequences map to particular secondary structures. 1,2 These authors have come to several remarkable conclusions, including the notion that most available secondary structures can be found in a relatively small subset of sequence space. It is therefore not necessary to explore the entire sequence space to attain the best (or evolutionarily successful) structure. Such findings have important implications and may be tested experimentally.
In vitro selection or directed evolution of nucleic acids involves many of the same steps that are operant during natural selection. In vitro selection of aptamers is accomplished with DNA or RNA molecules that have been selected from random pools based on their ability to bind other molecules. 3,4 We have previously selected for aptamers that bound to the protein lysozyme and found that a single binding sequence largely dominated the population. 5 A small number of other species were also isolated, and their relation to the dominant sequence is shown in Figure 1. The sequences were compared to each other using their pairwise Hamming distances. Hamming distance is a method of sequence comparison that quantifies the dissimilarity between two oligomers by tallying up the number of nucleotide mismatches in their optimal alignment. 6 It is possible that this binding sequence represented a “global optimum” for the selection. However, precisely because the selection experiment generated a fixed species, it was unclear whether the evolutionary landscape that connected nucleic acid sequences with lysozyme-binding function contained numerous, discrete local optima, or whether the landscape was similar to the “Mount Fujiyama” selection function described by Kauffman, 7 in that the closer sequences moved towards the global optimum, the better they bound to lysozyme. More data is essential to populate the sequence space and determine the structure for functional sequence fitness landscapes by investigating how these sequences pan out during in vitro selections. Greater number-selection experiments are essential to elucidate the evolutionary pathways that the anti-lysozyme aptamers take in order to attain their final functionality. By probing sequence space with more data, we will be able to populate the fitness landscape, which will provide us with the first glimpse into an evolutionary landscape using real experimental data. As a first step towards experimentally probing the nature of evolutionary landscapes using the wet-lab approach, we started from several arbitrary sequences and selected for lysozyme-binding function. A random sequence population of nucleic acids was initially generated by a combination of chemical synthesis and enzymatic amplification. Traditional in vitro selection techniques would require weeks to months for one set of selections to be completed; hence, conducting the described selection experiments would prove to be unfeasible. To overcome this obstacle, a Biomek 2000 Laboratory Automation Workstation (Beckman Coulter, Fullerton, CA) has been outfitted to carry out multiple in vitro nucleic acid selections in parallel, yielding substantial amounts of data for theoretical studies in a short amount of time. On the workstation, this population was parsed for function; in the current instance, nucleic acids that can bind to an arbitrary target protein, lysozyme, were selected. After each round of selection, the selected nucleic acid binding species (also known as aptamers) were amplified by a combination of reverse transcription polymerase chain reaction (PCR) and in vitro transcription. Automated in vitro selections allowed the generation of considerable amounts of experimental data in a short amount of time, making the link between theoretical studies with real bench work data attainable.
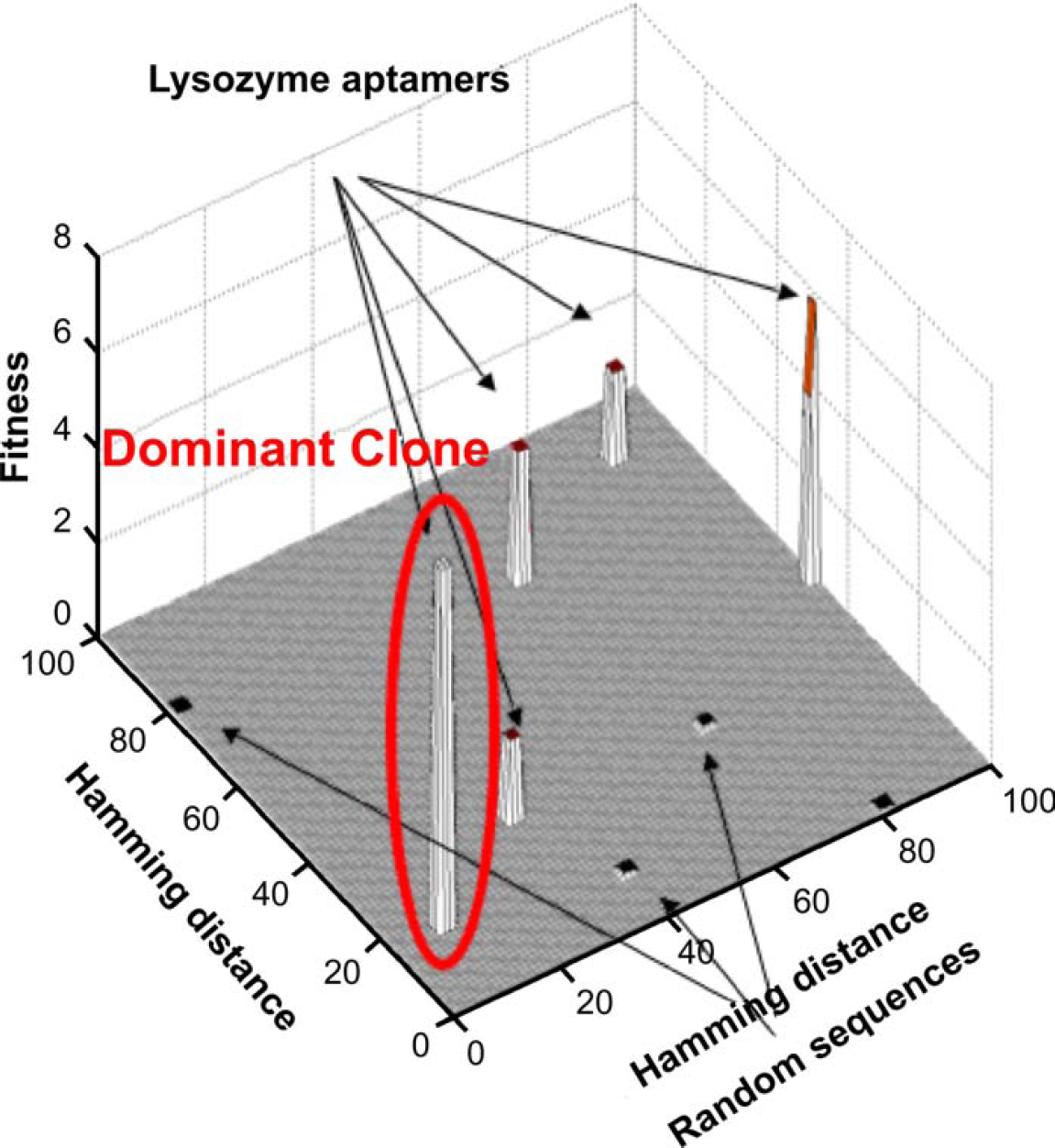
The isolated sequences were compared to each other based on their pairwise hamming distances. The fitness measure is based on the binding affinity of the species to the target. The dominant clone is highlighted in red and shows the highest level of fitness value as measured by their binding affinity to the target lysozyme.
Materials and Methods
Target Preparation
Hen egg white lysozyme from Sigma-Aldrich (St. Louis, MO) was chemically biotinylated using sulfo-NHS-LC-biotin (Pierce, Rockford, IL). Unincorporated biotin was removed via a 10DG chromatography-desalting column (Bio-Rad, Hercules, CA).
Library Generation
A single-stranded DNA library containing 40 random nucleotides flanked by two static priming regions was generated through chemical synthesis, which produced 1 × 1015 unique sequences. The entire library was then split into 8 aliquots before further manipulations were carried out as shown in Figure 2. Each aliquot (or genome) was amplified and transcribed separately to ensure its integrity and to prevent the possibility of cross contamination among the different pools.
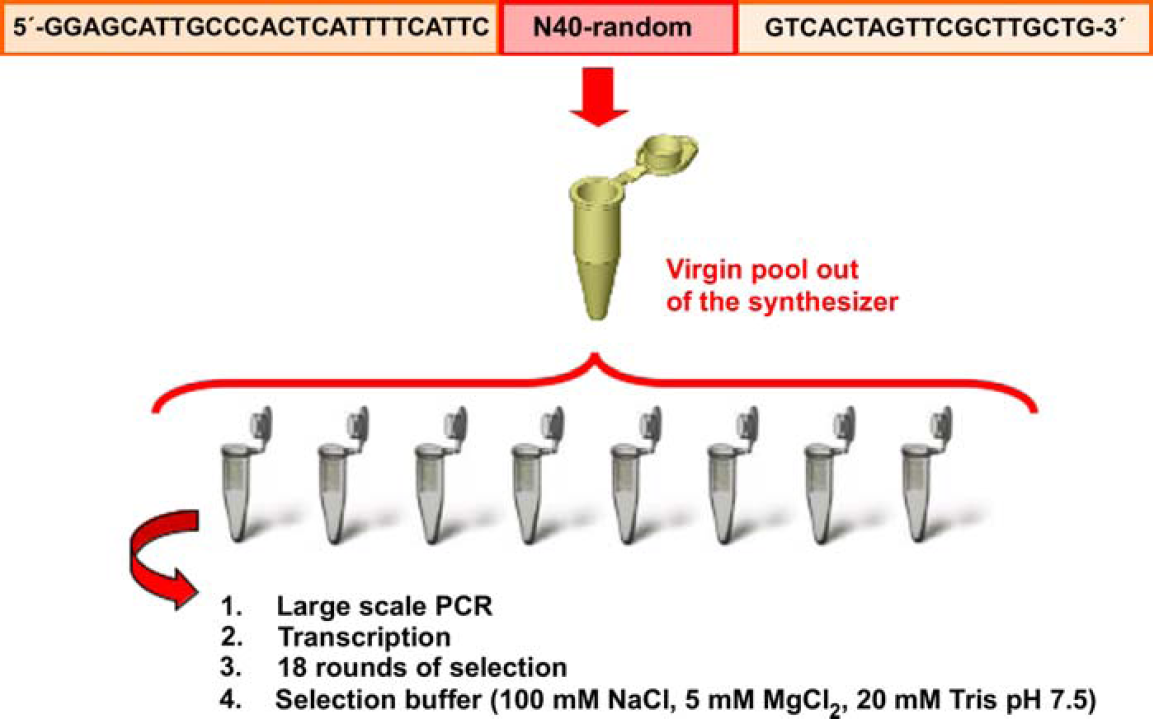
Initial pool is parsed into 8 different aliquots prior to the amplification process to ensure their integrity.
Selection Process
The eight RNA pools were carried through 18 rounds of in vitro selection against lysozyme in parallel using the Beckman Biomek 2000 workstation. The general schema for the selections is shown in Figure 3A. 5 To begin the selections, one genome of the RNA pool was incubated with the biotinylated lysozyme in selection buffer (20 mM Tris (pH 7.5), 100 mM NaCl, 5 mM MgCl2). Streptavidin-derivatized Dynabeads (Dynal Biotech, Brown Deer, WI) were used to “rescue,” or conjugate, the biotin-lysozyme and any bound RNA. The bead, target, and RNA mixture was then filtered with a Millipore HV (PVDF) filter on a vacuum manifold to partition the lysozyme binding RNA from the nonbinding RNA.
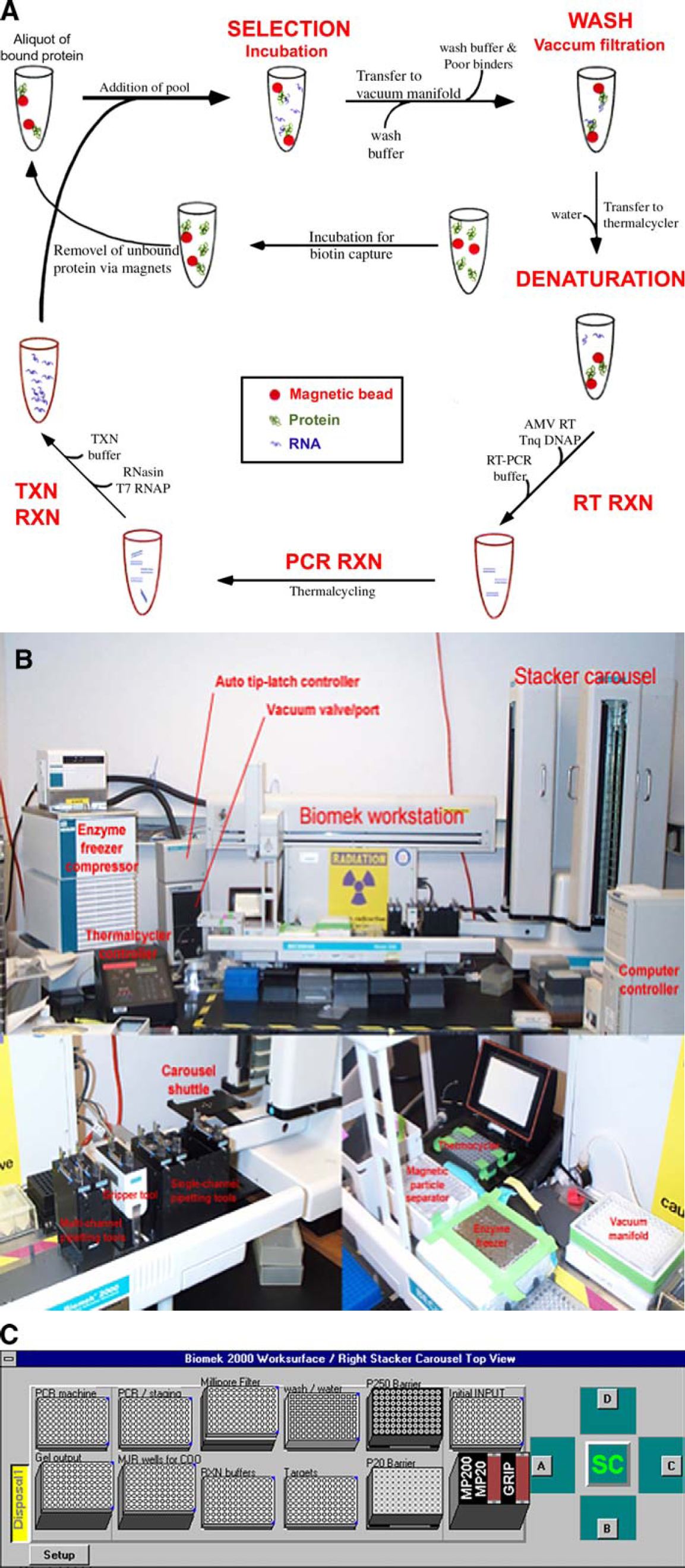
General selection setup. (A) The general schema for an automated selection. (B) The essential external components that are outfitted on the Biomek 2000 workstation to carry out automated selections. (C) The worksurface layout.
In the first round of selection, the amount of RNA pool applied was 5 mg (ca. 1.1 × 1014 sequences). Thereafter, approximately one-fifth of the preceding RNA transcription reaction was applied to the solution of biotin-lysozyme to begin the next round of selection.
The binding RNA was then amplified through a reverse transcription coupled PCR (RT- PCR) reaction followed by a transcription reaction. The reverse transcription coupled PCR (RT-PCR) reaction buffer contained 10 mM Tris (pH 8.4), 50 mM KCl, 1.5 mM MgCl2, 0.2 mM deoxynucleotide-triphosphates (dNTPs) 5% acetamide, 0.05% Nonidet P40, and 0.5 mM each of the primers). RT-PCR enzyme mixture contained 5 U of AMV reverse transcriptase (Amersham Pharmacia Biotech, Arlington Heights, IL), and 0.2 U of Display Taq (Display Systems, Vista, CA), 50% glycerol, 10 mM Tris (pH 8.4), 50 mM KCl, and 1.5 mM MgCl2. Binding species were eluted from the lysozyme by holding the mixture at high temperature.
A fraction of the RT-PCR mixture was used as template for the following transcription reaction to generate RNA for the next round of selection. The transcription buffer contained 40 mM Tris (pH 7.9), 26 mM MgCl2, 5 mM dithiothreitol (DTT), and 2.5 mM of each NTP. The transcription enzyme mix contained 40 U of RNasin (Promega, Madison, WI) ribonuclease inhibitor and 100 U of T7 RNA polymerase (Stratagene, La Jolla, CA).
Selection Worksurface
The selections was carried out on a Biomek 2000 Laboratory Automation Workstation, shown in Figure 3B. The robot work surface is integrated with a PTC-200 thermal cycler (MJ Research, Waltham, MA), a multiscreen vacuum filtration manifold (Millipore, Bedford, MA), and an enzyme cooler engineered in the laboratory as shown in Figure 3C. A stacker carousel (Beckman Coulter) feeds fresh pipette tips to the workstation as needed.
RNA Pool Assays
Individual pools were assayed for binding activity using a Minifold I filtration manifold (Schleicher & Schuell, Keene, NH) that sandwiched a Protran pure nitrocellulose membrane (Schleicher & Schuell) and a Hybond-N+ nylon transfer membrane (Amersham Pharmacia Biotech). Binding reactions were filtered through the filtration manifold and washed with the selection buffer. Percentages of bound nucleic acids were computed using a PhosphorImager SI (Amersham Pharmacia Biotech).
DNA generated from the selections was ligated into a thymidine-overhang vector using a TA Cloning Kit (Invitrogen, Carlsbad, CA). The transformants were grown overnight in Luria-BertaniMedia (LB) and DNA was isolated through direct colony PCR and cleaned up using PCR cleaning plates (Millipore, Bedford, MA). To obtain the sequence of aptamers, the cleaned PCR products were cycle-sequenced using a “sequence” reagent mix compatible with the Beckman CEQ™ 2000 sequences (Beckman, Fullerton, CA).
Results and Conclusions
A single robotic run on the Biomek 2000 Workstation performs six continuous rounds of aptamer selections in 18 h. The progress of each robotic run is assessed through the amplification of DNA and formation of RNA for each round of selection. This is observed by running aliquots of the reactions on a denaturing 8% polyacrylamide gel, such as that shown in Figure 4. The gel analysis shows whether the RT-PCR reaction was successful in yielding dsDNA and whether the transcription (TXN) reactions produced RNA.
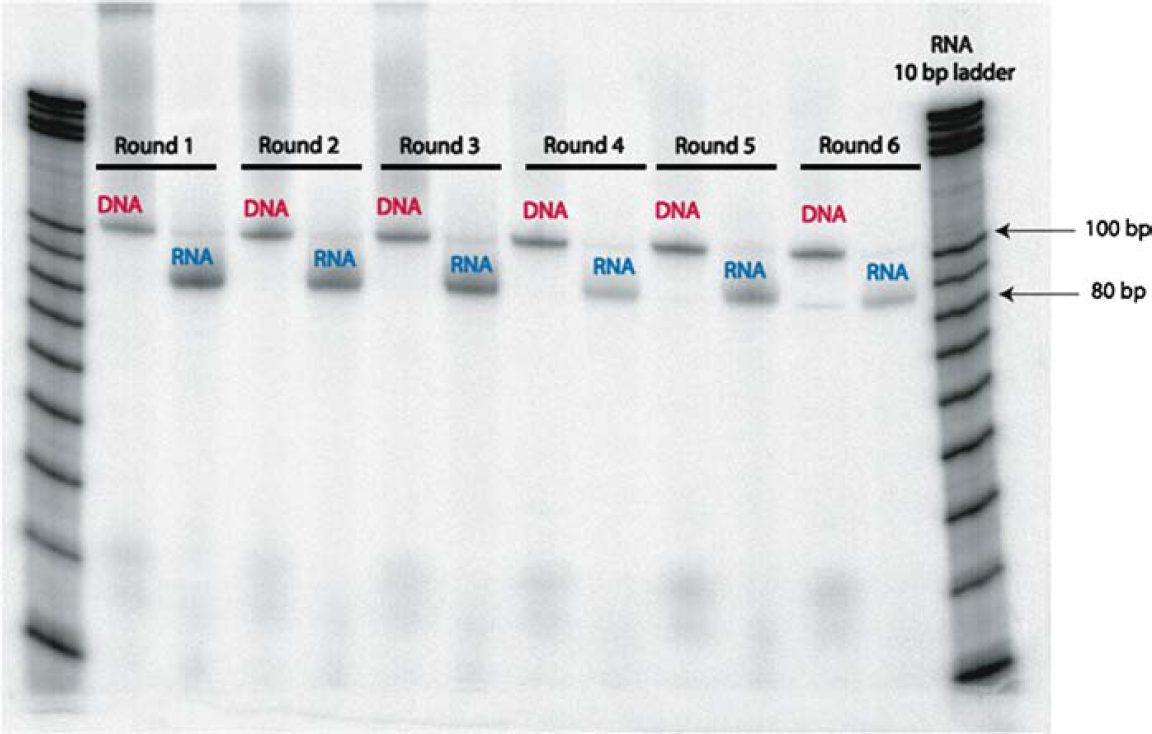
An analytical gel is used to ensure that both DNA and RNA are being generated at every round of the selection process.
Phylogenetic analysis of the round 18 sequences show that all eight different pool aliquots have been selected successfully without cross contamination across the different pool aliquots during the selection process as shown in Figure 5. Identical sequences within the same pool aliquot were input only once into the analysis. The phylogenetic analysis shows that no two sequences from the different pools shared the same branch, as it would be expected from identical sequences. The number of sequences was reduced in order to provide an intelligible graphical representation. Reducing cross contamination is one of the greatest challenges for automated selection. The lack of enclosure of the robotic platform makes contamination a likely event during the run. Thorough cleaning after each run and the use of filter systems that remove nucleic acid aerosols reduced this phenomenon. (Full sequences are available upon request.)
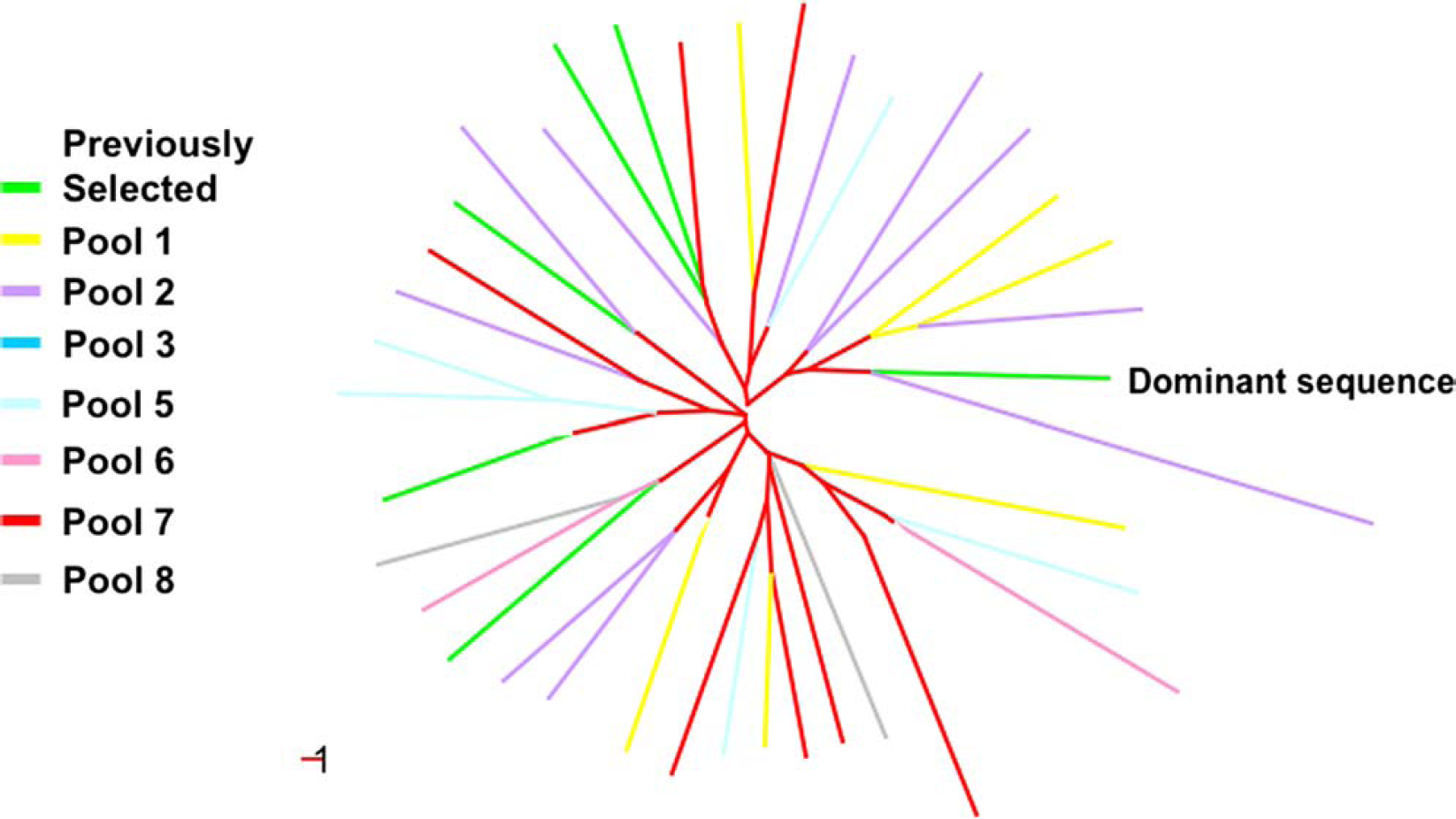
Phylogenetic analysis of the sequences isolated. The number of sequences used has been reduced in this graph. Previously isolated aptamers are incorporated into the analysis to show that no previously isolated aptamers have been isolated in the new selections.
The assays for the different pools were carried out on the same day to reduce the amount of variation during sample manipulations. The results also show very diverse binding trends across the different rounds of selection. Figure 6 shows the binding trends of four of the eight pools that have undergone selection. The level of binding affinity displayed by the pools was also very different. For instance, aptamers in Pool 1 show a steady increase in their affinity to the lysozyme as the selection progressed. Pool 2 shows that the affinity increased significantly from round 12 to round 18. Aptamers from Pool 3 appear to have their affinity fixed at round 12, since no significant difference in their binding percentage was observed between round 12 and round 18. Finally, Pool 4 binding affinity soared from round 6 to round 12 and decreased significantly at round 18. This implies that every pool, which contains different sequences each, has undergone different evolutionary pathways to attain their functionality, hence the existence of such a variety of binding trends. If the pools were to be composed of closely related or similar aptamer sequences, such a wide range of binding percentage would not be expected among different pools. Clone 1 used in the assay was the previously selected lysozyme aptamer 5 serving as the positive control. RNA was also incubated with the Dynalbeads and assayed to ensure that the aptamers selected were not bead binders.
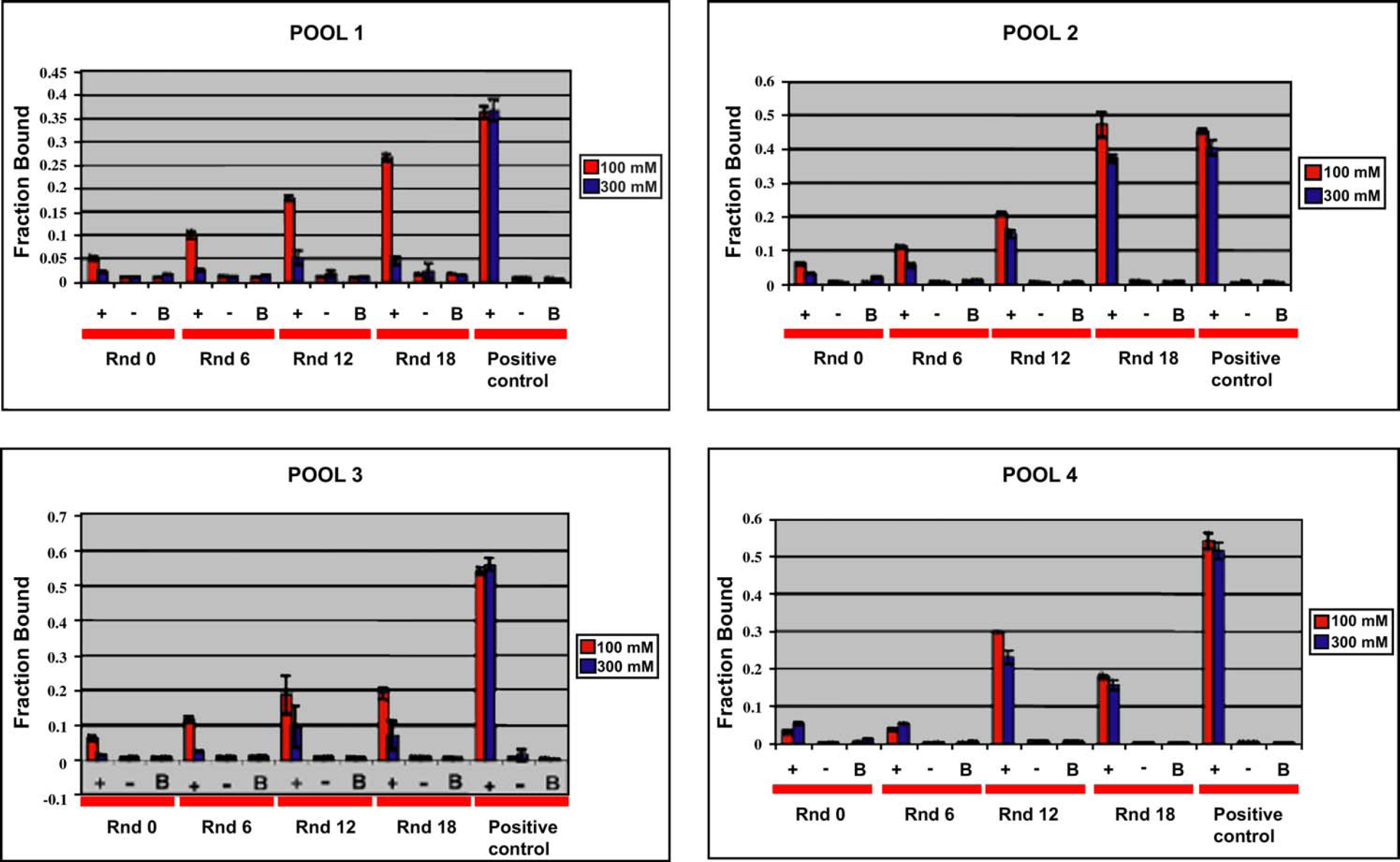
Binding assays of the selected pools. The selected RNA pool is incubated with the protein (+), without protein (−), and with beads (B). The assays are carried out using two different washes of different salt concentrations to reduce background binding. The figure depicts how different the binding trends are for the different pools.
In vitro selections are often carried out only once without partitioning the initial pool. The isolated binding aptamer is assumed to be the fittest among the population because it is the sole aptamer isolated. In this experiment, it can be inferred that when given a chance to evolve, other species can be isolated. The results also show that no two pools display the same behavior in terms of their evolutionary pattern for their binding traits. During in vitro selection, many different factors can prevent the isolation of the best binder. Some RNA aptamers that have better amplification capability might outcompete other more stable structures that are unable to amplify as efficiently. By splitting the pools before the process of selection, we are allowing more room for other species to win the competition, shedding light into whether the previously selected winner is indeed the globally optimal aptamer.
Individual isolated sequences will be further analyzed and screened for their affinity towards the selected target and compared to previously isolated aptamers. The results can shed light onto the future selection process by taking into account the different factors that can prevent the isolation of a better aptamer that might have been overlooked. With the advent of automated selection, it is now possible to conduct theoretical studies utilizing experimental data to attain a more accurate model.
Acknowledgments
This research is funded by the National Science Foundation's Integrative Graduate Education and Research Traineeship (IGERT) in computational phylogenetics. Special thanks to Gwen Stovall for her suggestions and comments.