
Abstract
Methods for the high-throughput identification of transcription factor binding sites (TFBSs) are becoming increasingly relevant as new genomes are sequenced and explored. Although DNA:protein complexes can be purified from lysates and binding sites identified, this only provides a snapshot of the full DNA-binding potential of an organismal proteome. Instead, automated double-stranded DNA (dsDNA) selections can be used to identify target sites for all DNA-binding proteins in parallel. Double-stranded DNA selections were carried out against cell lysates from Escherichia coli. The resultant aptamer sequences showed enrichment for known TFBSs versus both the native pool and the E. coli genome, and underwent an overall change in sequence composition. Randomized versions of the selected pools did not show the same enrichment. Based on this automated method and accompanying bioinformatics algorithms, it may be possible to extract complex information about TFBSs from cellular lysates. It may further be possible to use these methods to model previously unknown DNA-binding sites in a variety of novel organisms. (JALA 2007;12:135–42)
Background
There are few high-throughput methods for decon-voluting the many interactions between nucleic acids and proteins in cells. In one approach (so-called ChIP-chip methods),1, 2 proteins are cross-linked to DNA, the proteins are immunoprecipitated, and the DNA fragments are identified using microarrays. However, this process is relatively laborious, only assesses a single state of the cell, and can only be applied to one transcription factor or other protein at a time.
Selection experiments can also be used to identify nucleic acid:protein interactions, and can potentially be automated and carried out in a high-throughput fashion. For example, both automated and manual selections against the NFkB p50 homodimer have yielded the in vivo binding site.3, 4 Similarly, Benbrook and Jones performed selections against CREB1, CREB2, and the CREB2/cJun heterodimer. 5 All of the selected sequences were similar to the known decamer binding site. Selections have also been used to successfully identify unknown binding sites, for example, the HspR binding site in Helicobacter pylori. 6 Newly identified binding sequences were later confirmed by DNA footprinting. It should be noted, though, that in vitro selections can sometimes yield binding sequences that differ from those in vivo. 7 Shultzaberger and Schneider found that in vitro selection experiments identified a binding site for a dimeric or trimeric form of Lrp, whereas in fact Lrp acts as a monomer in vivo and binds an alternate site. To avoid such discrepancies, it may be useful to carry out selections in an environment that is as similar to the target's native environment as possible, such as a cellular lysate.
Selection methods have previously been applied in whole cell lysates. For example, antibody selections have previously been carried out against cell lysates8, 9 and have yielded antibodies specific for the antigens within the lysate. Nucleic acid selections against cell lysates have also been performed.5,10,11 Often a rabbit reticulocyte lysate, as prepared by Jackson and Hunt, 12 is used to overexpress a protein of interest. For example, when Weintraub and coworkers11, 13 performed selections against MyoD and E2A homo- and heterodimers the natural consensus sequence CANNTG was identified, in addition to protein-specific variations of this motif. Selections that targeted purified myogenin and myogenin in nuclear extracts have also produced the known in vivo binding site.14, 15
Based on these results, it should also be possible to select against lysates to gauge the DNA-binding capacity of the lysate as a whole, as opposed to identifying single binding sites. By using robotic selection procedures, it should be possible to improve the surety and reproducibility of multiple site identification, as well as providing capacity to look at multiple different lysates from multiple organisms or variants in a high-throughput manner. In addition, these proof-of-principle experiments provided an opportunity to test the versatility of the Tecan Genesis robotics platform at performing multiple molecular biology tasks.
Materials and Methods
Liquid Handling Robot
A Tecan Genesis workstation 200 was used to automate the selection of dsDNA molecules that could bind to proteins present in cellular lysates. The Tecan has both a liquid handling pod and a pod containing a robotic manipulator arm. The liquid handling pod contains eight independently controlled pipette tips that are capable of pipetting between 0.5 and 1000 μL, whereas the RoMa arm can reach off of the worksurface. Using the arm, additional equipment can be integrated with the Tecan workstation.
The Tecan Genesis worksurface holds a variety of equipment (all from Tecan, unless otherwise indicated), including a twelve position microplate carrier (MP-12), a solid-phase extraction unit with an adapter for Qiagen (Valencia, CA) kits, a two position orbital shaker, a 4 °C cooled microplate carrier with a recirculating temperature bath which holds 1.5 mL tubes (Julabo, Allentown, PA), disposable tips, and reservoirs for other reagents. Items off the worksurface but accessible by the RoMa arm include a thermal cycler (MJ Research, Waltham, MA) and a Tecan 16-channel Columbus plate washer.
Oligonucleotides
The LS.N65 pool contains N56 between a 5′ constant region (5′ GATAATACGACTCACTATAGCTTA) and a 3′ constant region (5′ ACGTCTCGTCAAGTCTGCAATGTA). Some 10 13 synthetic DNA molecules were amplified in a 5 mL PCR reaction over six cycles. Following amplification, the double-stranded N56 pool was purified using a Qiagen PCR clean up kit (Qiagen, Valencia, CA). A fraction of the purified pool amounting to 10 13 amplified molecules was used in the initial round of the selection.
Target Plate Preparation
Cell lysates were prepared from two different cell lines. The first was BL21(DE3) cells from Novagen (San Diego, CA) containing pACYC(CAM), a plasmid that contained an expression cassette for LacI (Novagen, San Diego, CA). The second was BL21(DE3) cells from Novagen containing pASK-IBA3, a plasmid that contained an expression cassette for TetR (IBA, St. Louis, MO). One liter cultures containing 20 μg/mL of chloramphenicol were grown to saturation and then spun down in 500 mL culture bottles at 4000 g for 15 min. The pellets were washed with 5 mL of SXN and spun down again. Then, the pellets were transferred with a metal spatula to 50 mL conical tubes where 5 mL of SXN and 800 μL of Roche Complete protease inhibitor cocktail (Roche, Indianapolis, IN) were added. Each pellet was resuspended on ice. Cells were disrupted with alternating 10-s intervals of sonication (at 13% on a Fisher Scientific Sonic Dismembrator Model 500 [Pittsburgh, PA]) and resting for a total of 20 min. Following sonication, the lysates were spun at 10,000 g for 45 min. The supernatant was removed and used to prepare the target plates.
Cell lysates were loaded into wells of TopYield microtitre plates (Nunc, Rochester, NY). Some 100 μL of lysate and 200 μL of SXN were added to each well. The wells were sealed with a microplate seal, and the plates were incubated without agitation at 4 °C for approximately 18 h to allow hydrophobic bonding of proteins to the surfaces of the wells.
Automated Selection
The selection process as a whole is diagramed in Figure 1 and the details of individual selection cycles are provided in Table 1. The target plate was placed on the MP-12 on the Tecan worksurface. The lysate solution was removed from the wells of the plate and the wells were rinsed with 175 μL of SXN. The Round 0 dsDNA pool (100 μL; 1.5 μg; 10 13 sequences) was transferred from the 4 °C cooled microplate carrier to the selection plate. The selection plate was then transferred to the orbital shaker where it was iteratively rotated for 3 min at 500 rpm and then allowed to stand for 5 min (see Table 1 for the number of cycles). The plate was then moved to the Columbus plate washer and wells were washed with varying numbers (see Table 1) of 175 μL aliquots of SXN and dH2O. The target plate was moved back to the MP-12, and PCR master mix (100 μL; 10 mM Tris, pH 8.4; 50 mM KCl; 2.5 mM MgCl2; 0.2 mM dNTPs; 0.4 μM each of the 5.24.N56 5′ primer and the 3.24.N56 3′ primer) and 5U Taq polymerase were added. The target plate was transferred to the thermal cycler for PCR amplification (denaturation for 10 min at 90 °C, then cycled for 90 s at 90 °C, 30 s at 49 °C, and 90 s at 72 °C; final extension for 3 min at 72 °C; number of cycles as in Table 1). During the thermal cycling procedure, the Columbus aspiration and dispensing needles were cleaned with 6 mL of a 7 M urea solution followed by 6 mL of dH2O. Following DNA amplification, the program was paused and a 4% agarose gel was run to make sure that dsDNA had been produced and that amplification artifacts had not accumulated.
Selection conditions and stringency
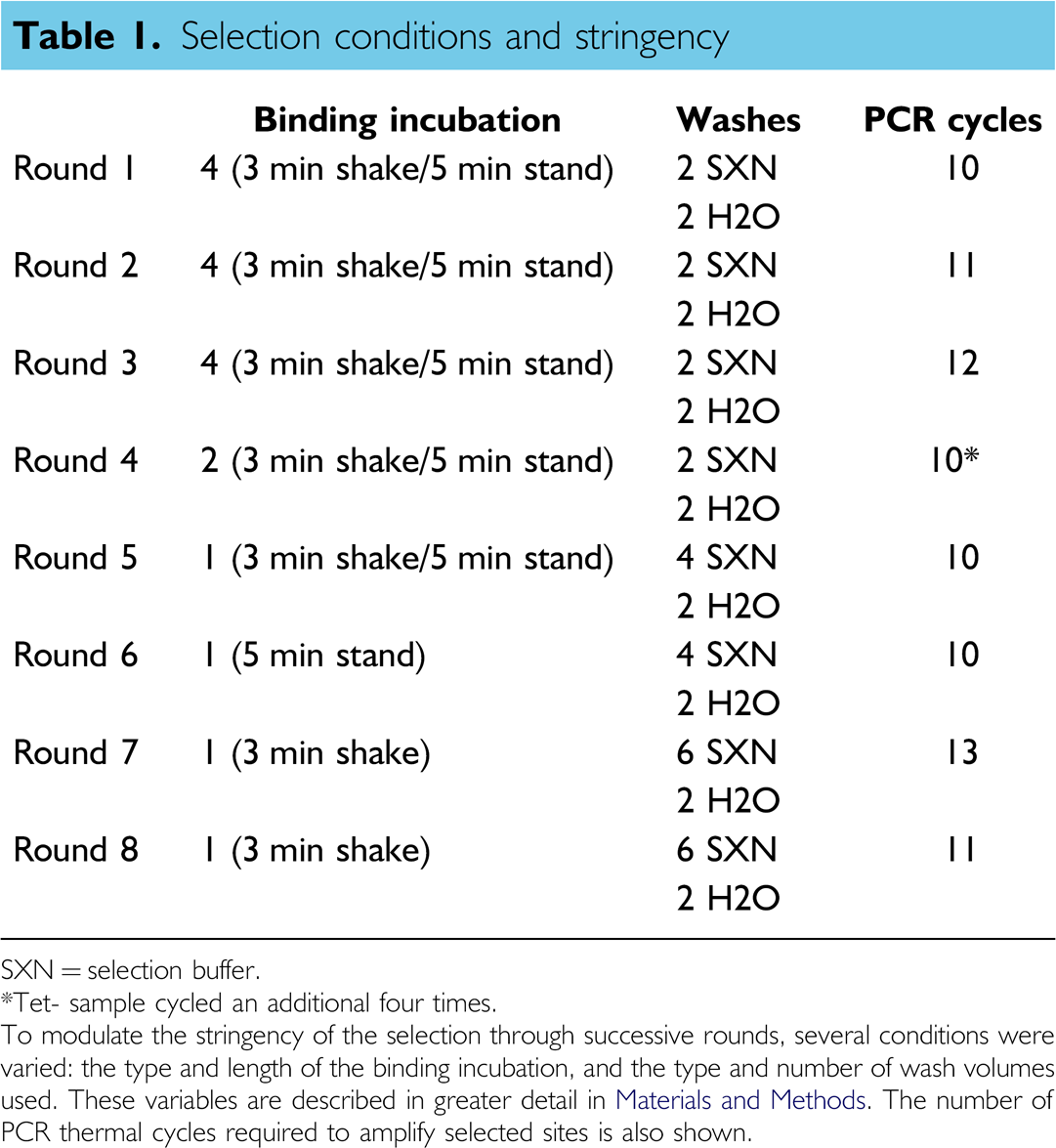
SXN = selection buffer.
Tet- sample cycled an additional four times.
To modulate the stringency of the selection through successive rounds, several conditions were varied: the type and length of the binding incubation, and the type and number of wash volumes used. These variables are described in greater detail in Materials and Methods. The number of PCR thermal cycles required to amplify selected sites is also shown.
Automated panning protocol for double-stranded DNA (dsDNA) selections. This figure is a simple schematic of the protocol described in Materials and Methods. In short, a dsDNA library (dual lines) was transferred to a microtitre plate well containing target lysate (grey circles). Loosely or nonspecifically bound DNA species were washed away. A PCR reaction mixture was added directly to the well and any remaining DNA molecules were amplified. The amplified products were purified and then added to a new selection well to begin the next cycle of selection and amplification.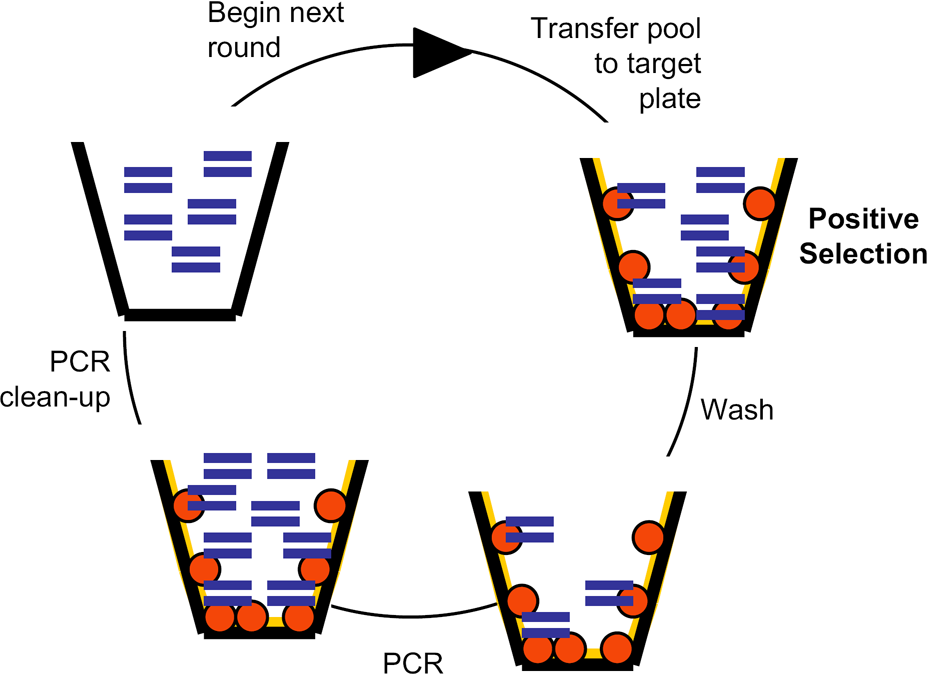
Following quality control by electrophoresis, the plate was returned to the 12 microplate carrier on the worksurface and 15 μL 3 M sodium acetate (pH 5.2) was added to the wells to lower the pH of the PCR reaction to pH 6–7 (a more optimal pH range for the subsequent Qiagen clean-up kit). The acidified reaction mix was added to 345 μL Qiagen Buffer PM in a 2 mL deep-well plate on the MP-12 worksurface. The mixture was transferred to the Qiagen filter plate on the solid-phase extraction device. A 500 mbar vacuum was applied for 5 min to pull the solution through the filter on the Qiagen plate. Then, 900 μL of Qiagen Buffer PE was added, followed again by application of a vacuum. The final wash was an addition of 900 μL of Buffer PE. The filter was dried under vacuum for 10 min. For the collection of the DNA eluate, 120 μL of SXN was added to the well, the robotic manipulator arm transferred the solid-phase extraction device to a second position on the manifold, and a vacuum of 500 mbar was applied for 5 min. The purified PCR product was collected in a Qiagen deep-well plate, and the solid-phase extraction device was transferred back to the first position on the manifold by the robotic manipulator arm for use in the next cycle of selection. The final 100 μL of eluted, dsDNA was transferred from the deep-well plate position on the vacuum manifold to a freshly washed, lysate-coated microtitre plate to begin the next round of selection and amplification.
Sequencing
A standard automated sequencing protocol was used to sequence the selected pools. The dsDNA pools from Rounds 0, 5, and 8 were cloned into TOPO TA vectors (Invitrogen, Carlsbad, CA) and transformed into Top 10 (Invitrogen) competent cells. Following transformation, cells were plated on Luria-Bertoni media (LB) plates supplemented with 50 μg/mL kanamycin and 1600 μg X-gal per plate. The plates were incubated at 37 °C until small colonies were visible. White colonies were picked and used to inoculate 1 mL cultures of LB containing 50 μg/mL ampicillin in a 2 mL 96-well deep-well plate (Corning, Acton, MA). Cell cultures were grown overnight at 37 °C with shaking, and 2 μL of cells were used directly as templates for 100 μL PCR reactions. The 2 μL aliquots of cells were first boiled at 100 °C in 78 μL dH2O for 10 min, then 19 μL of PCR master mix (final concentrations 10 mM Tris, pH 8.4; 50 mM KCl; 2.5 mM MgCl2; 0.2 mM dNTPs; 0.4 μM each of the M13(-40)F and M13R primers), and 1 μL (5U) of Taq polymerase were added. Following 15 thermal cycles (denaturation for 3 min at 95 °C, then cycled for 45 s at 95 °C, 30 s at 45 °C, and 90 s at 72 °C; final extension for 3 min at 72 °C), PCR products were purified with a Millipore (Billerica, MA) PCR clean-up kit and sequenced with Big Dye v3.0 mix (ABI, Foster City, CA). Sequencing reactions were analyzed on an ABI 3700 automated sequencer.
Sequence Analysis
The selected sequences from each of the five pools were analyzed using an application written in Perl. The program takes as input the sequences from each pool and a set of TFBS. For data analysis, the TFBS set was that of known E. coli transcription factor binding sequences, obtained online from RegulonDB (http://www.cifn.unam.mx/Computational_Genomics/regulondb/). 17 Sets of randomized sequences were used as controls. Each set of randomized sequences contained the same number of sites of each length class as the known set of binding sites (e.g., thirty 20-mer sites) while the base contents varied. One set matched the base content of the Round 0 pool (A 30.66%, C 26.06%, G 19.48%, T 23.80%), whereas a second randomized set contained equimolar amounts of the four bases.
Each binding site was then broken into hexamer registers for both the forward and reverse strands. The program looked for each register within all of the aptamers in each pool. When a match was found, the script extracted a sequence from the aptamer, which corresponded to the alignment between the TFBS and the aptamer, anchored at the identified hexamer (see also Fig. 3). Once the transcription factor-like sequences were extracted from the aptamers, the program was then able to tabulate informative statistics, such as the number of times each pool showed better similarity to a known sequence relative to a randomized sequence.
Another Perl script was developed to analyze the sequences using a log likelihood approach. Such an approach had previously been used to identify Medline abstracts that discussed protein–protein interactions.
18
Briefly, the program first determined the frequencies of individual hexamers in the set of known TFBSs in the RegulonDB, the entire E. coli K-12 genome from Genbank, and each of the aptamer pools. The program then calculated a score for the frequency ni for a given hexamer i in the aptamer pool with total hexamers N relative to that hexamer's frequency in the database of binding sites fI, i and the genome fN,i. By summing these scores for all the hexamers in the known TFBS, we calculated a log likelihood score S for the whole pool:
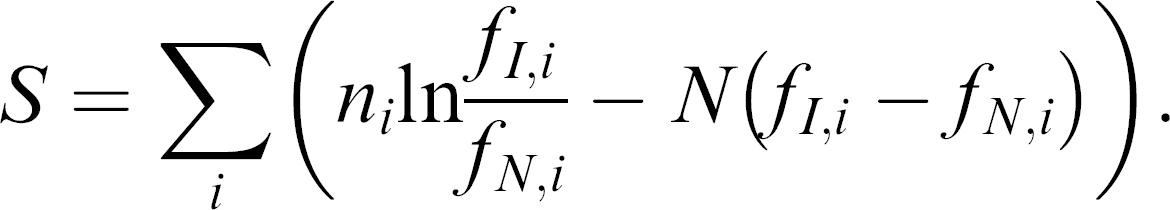
Results and Discussion
Selection of Double-Stranded DNA Aptamers That Bind to Cell Lysates
Selections were performed using a double-stranded DNA (dsDNA) pool that spanned fifty-six random residues (N56) flanked by two constant priming regions. This pool was allowed to bind to lysates from two different derivatives of the same parental Escherichia coli strain. Both derivatives were transformants of the common laboratory cloning strain BL21(DE3) (Novagen, San Diego, CA). One derivative contained the pACYC(CAM) plasmid (Novagen, San Diego, CA) and overexpressed the LacI protein, whereas the other derivative contained the plasmid pASK-IBA3 (IBA, St. Louis, MO) and overexpressed the TetR protein. The two different strains served as controls for one another, and allowed us to determine whether and how the presence of particular DNA-binding proteins might skew the selection of aptamers.
The selection procedure itself was relatively straightforward (Fig. 1), but still required a number of adaptations for full automation. For a highly detailed explanation of the automated selection process, please refer to Materials and Methods: Automated Selection. First, E. coli lysates were incubated in microplates composed of nonpolar polymers. Many proteins have numerous nonpolar residues on their surfaces, and therefore proteins within the lysate bound to the microplate wall through hydrophobic interactions. This method of target preparation is commonly used to immobilize proteins for ELISA and has previously been successfully used in an automated selection that targeted the NFkB p50 homodimer. 4 Second, the microplate was placed on the worksurface of a Tecan Genesis workstation for robotic manipulation. The wells were rinsed with selection buffer (SXN) to remove any weakly bound proteins. The double-stranded N56 DNA pool (10 13 species) was incubated in the wells. Double-stranded DNAs that bound to the immobilized lysate remained behind when the plate was washed with 1.8–5.6 mL of SXN and 600 μL of water. The remaining, captured dsDNA-binding species were then amplified via the polymerase chain reaction (PCR). Following amplification, the robot was paused while a 4% agarose gel was run directly on the worksurface to check for the presence of dsDNA products of the appropriate size, and to make sure that artifactual products did not accumulate. This ability to integrate selection, amplification, and product analysis was critical to the success of the protocol, and could only be automated on a complex, multitasking workstation. Following confirmation that a band of the correct size had been obtained, the PCR product was purified (again on the robot's worksurface) using a Qiagen PCR clean-up kit. The newly amplified dsDNA was then used for additional rounds of selection and amplification.
The overall stringency of the selection was low compared with previous selections against individual dsDNA-binding proteins. However, the stringency of the selection was progressively increased during the selection, to narrow the selection to a defined group of binding sequences. The time allowed for binding was decreased while the number of wash cycles before elution was increased (Table 1). Selected DNA pools were recovered after five and eight rounds of selection. The pools were then sequenced. Some 30 sequences were obtained from Round 0, while 85 additional sequences were obtained from Rounds 5 and 8 of the selection targeting LacI expression, and 77 additional sequences were obtained from Rounds 5 and 8 of the selection targeting TetR expression.
Analysis of Selected Sequences
Comparison of Selected and Unselected Sequences. It seemed unlikely that the selected binding sites would be perfect replicas of genomic DNA-binding sites for several reasons. First, there is often great diversity between the natural DNA-binding sites of a given transcription factor or DNA-binding protein. In line with this observation, previous selection experiments had shown that dsDNA molecules selected to bind individual DNA-binding proteins yielded known consensus sequences but did not perfectly mimic genomic binding sites.3,4,7 For example, Kunsch and coworkers selected binding sites for p50 homodimers that yielded the known consensus GGGRNNYYCC, but most sequences did not match the strongest in vivo binding sequence, GGGGATTCCC. Second, the conditions for the selection that targeted lysates were not particularly stringent, compared with previous selections that targeted individual DNA-binding proteins. Relatively nonstringent conditions were chosen to target multiple DNA-binding proteins in the lysate, including proteins that were relatively rare or had low-binding affinities. However, these conditions also likely allowed many, noncanonical variants of higher affinity binding sites to be retained in the population. Finally, given that each selected aptamer would have been exposed to a variety of targets during subsequent rounds of selection, those sequences that could bind to multiple, different targets might ultimately predominate in the population. Such chimeric binding sequences would not necessarily resemble a single, genomic binding site.
For these reasons, our initial measure of success was the analysis of whether short sequences within the selected molecules were found in the known set of E. coli transcription factor binding sites (TFBSs), and whether this was greater in the selected pools versus the unselected pool. To identify homology between pools and TFBSs, we first enumerated all individual hexamer sequences from known TFBSs within either selected or unselected pools.
The selected pool contained many more hexamer binding sequences than did the unselected sequence pool; this can best be seen by looking at the normalized distribution of hexamers across different sizes of transcription factors (Fig. 2). Although there were some differences between the selected sequences from Round 5 to Round 8, and between sequences selected from lysates that contained either LacI or TetR, these differences were much less significant than the overall differences between selected pools and unselected pools. Moreover, irrespective of whether small or large TFBSs were examined, there were many more hexamer binding sequences in the selected pools than in the random pools. This conclusion proved to be true irrespective of the length of sequence chosen for analysis, but can be most readily seen for hexamers.
Distribution of selected hexamers also found in transcription factor binding sites (TFBSs) across TFBSs of different lengths. This plot shows the number of hexamers found in TFBSs of different lengths relative to a given pool. The number of matches differs for TFBSs of different lengths primarily because there are different numbers of TFBSs of different lengths (i.e., more 22-mer binding sites than 23-mer binding sites). All four selected pools show a clear enrichment in hexamers found in TFBSs over the random (Round 0) pool.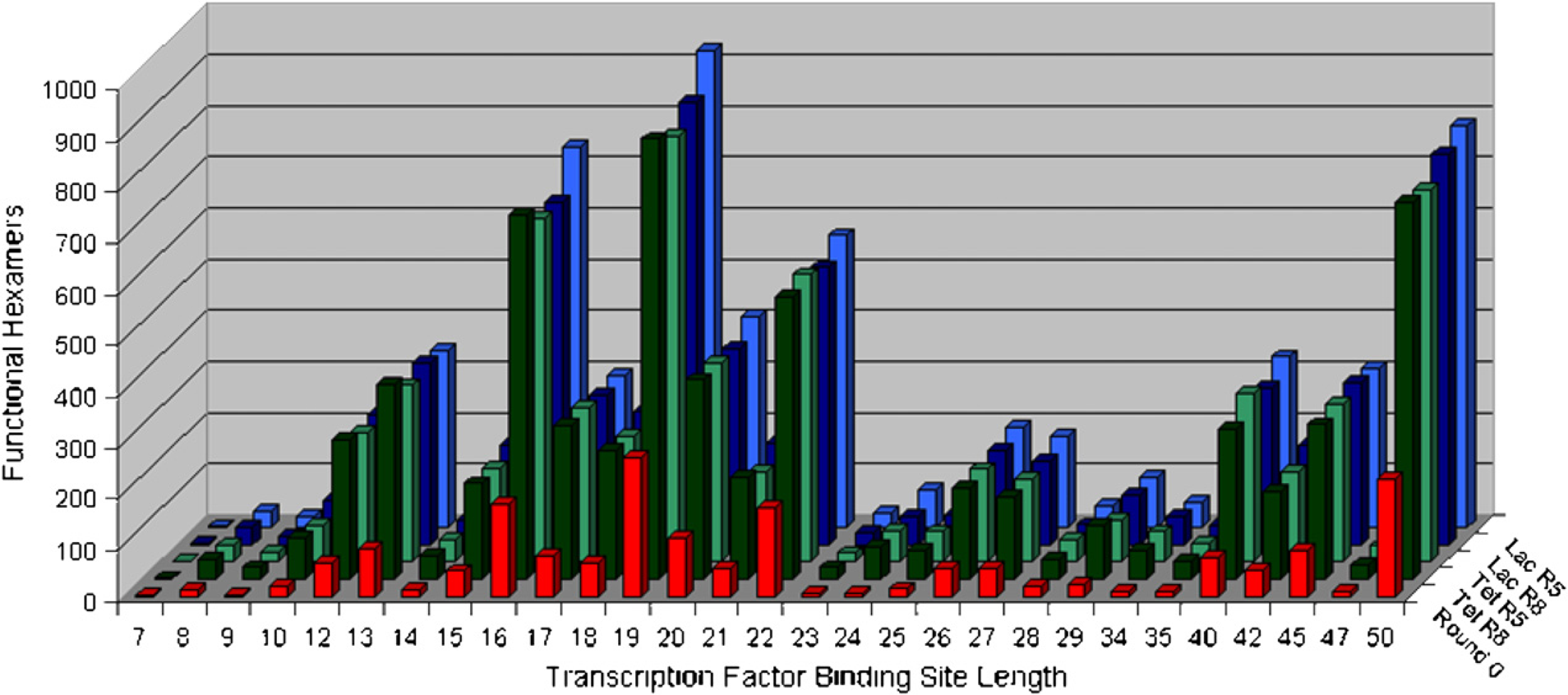
The preponderance of known binding sites in the selected pools was also apparent when overall sequence similarities rather than hexamer sequences were examined. As before, the E. coli TFBS database was used as a source of known binding site hexamers, but instead of just comparing these hexamers and those in the selected pool, the known binding site hexamers were used to “anchor” alignments between the database and the selected aptamers (Fig. 3). The hexamer-anchored alignments were then scored in terms of overall similarity. As a negative control, a randomized version of the E. coli database was used. The similarity values obtained with either selected or randomized databases were similar, but the selected aptamers were always slightly more similar to the actual TFBSs than the randomized sites (data not shown).
Analysis of selected sequences for transcription factor binding sites (TFBSs). (A) Both the forward and reverse strands of known Escherichia coli TFBSs were broken into hexamer “words.” (B) Each word was checked to determine whether it was found in a selected aptamer. (C) Once a match was found, additional sequences outside of the hexamer word (light grey) were then compared to the known TFBS. (D) The overall alignment outside of the hexamer word was scored. In this case, the extracted sequence matches 9 out of 15 bases for a score of 0.6. These scores were used to compare different pools. 1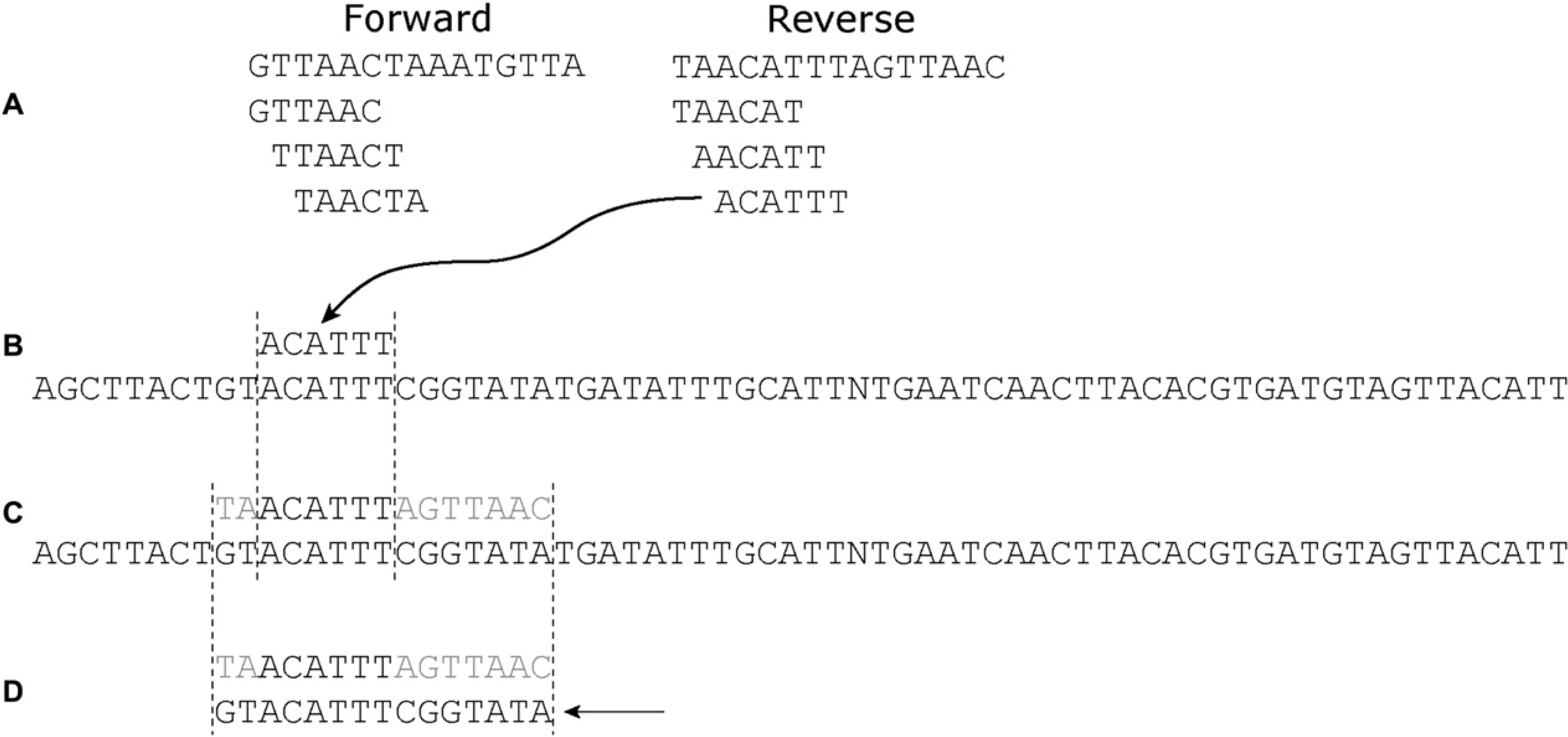
The small skewing that was observed was less than that might have been expected based on the analysis of hexamers as shown in Figure 2, where there were apparently many more binding site sequences in the selected sequence population relative to Round 0. However, closer analysis resolves this apparent contradiction. The similarity of selected aptamers to TFBSs (randomized or not) was greater than the similarity of Round 0 sequences to TFBSs (randomized or not). The most likely explanation for this result is that the sequence composition of the population was under selection, and thus even randomized sequences were more likely to match selected aptamers than the Round 0 population. This is in fact true (Table 2). When the sequence composition of the selected pools is compared with Round 0, the proportion of C residues decreases (from greater than 25% to less than 20%) and the proportion of T residues increases (from 20% to 30%). The selected populations almost exactly fit the average sequence composition of known E. coli TFBSs.
Nucleotide compositions of pools and transcription factor binding sites
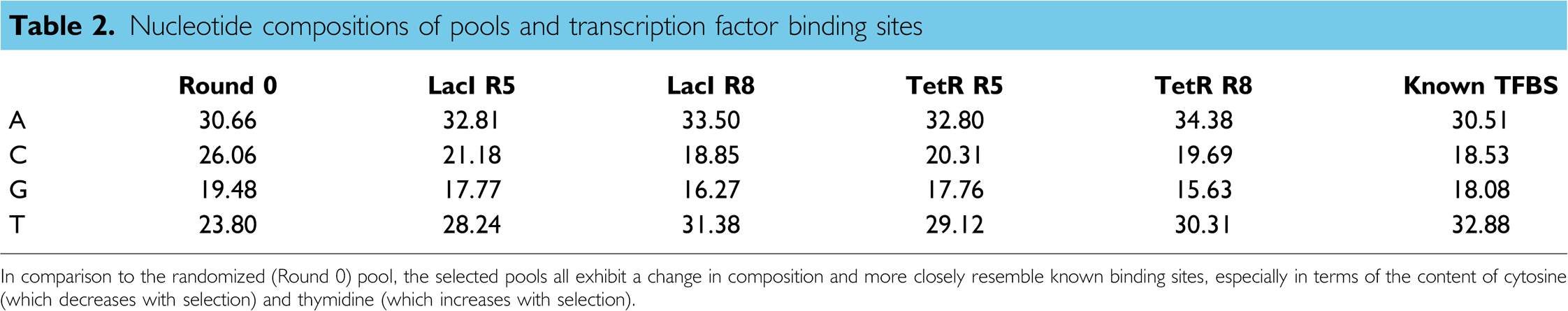
In comparison to the randomized (Round 0) pool, the selected pools all exhibit a change in composition and more closely resemble known binding sites, especially in terms of the content of cytosine (which decreases with selection) and thymidine (which increases with selection).
Anecdotally, there were a number of duplicated clones and sequences in Rounds 5 and 8 of each selection. These sequences tended to correspond to sequences that would be found in the -35 and -10 regions of promoters, or A Box transcriptional activator sequences. Interestingly, there was no apparent preference in any of the pools for particular TFBSs: the selected sequences that targeted the LacI-expressing cell lysate were as likely to contain TetR binding sites as were the selected sequences that targeted the TetR-expressing cell lysate. The selections appeared to yield a representative set of binding sequences for the many other transcription factors in the lysate.
Log likelihood scores for matches between transcription factor binding sites (TFBSs) and pools. Log likelihood scores were calculated as described in the text. A negative score evidenced that sequences in a given pool were skewed toward the Escherichia coli genome as a whole, whereas a positive score indicated that the sequences in the pool were skewed toward known TFBSs. The four bars represent comparisons with the variable region by itself (blue), the variable region plus three residues on either end (maroon), the variable region plus six residues on either end (grey), and the whole aptamer (green).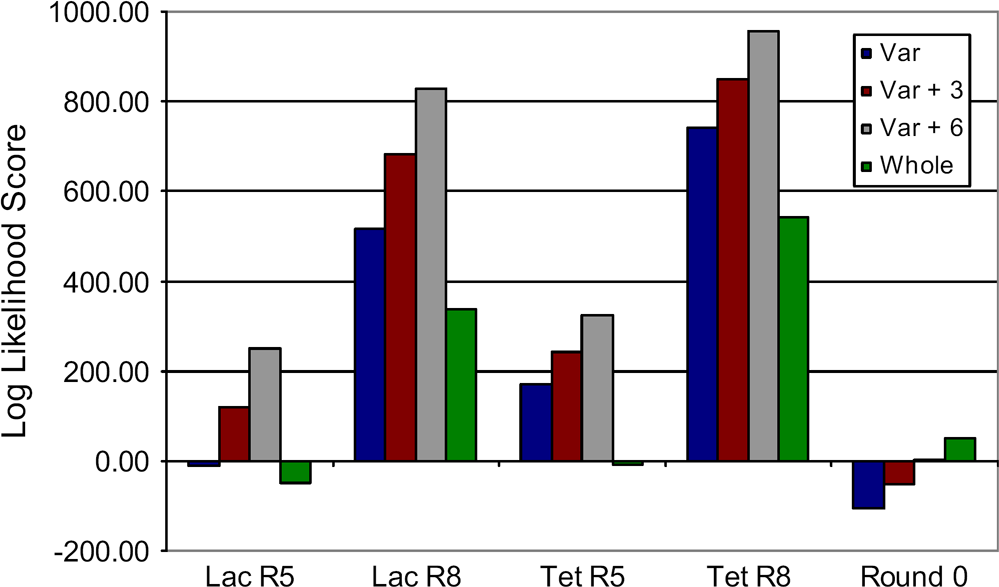
We also examined whether the constant and the random regions were potentially involved in binding to the transcription factors. These comparisons were made with (1) just the variable region, (2) the variable region plus three bases on either end, (3) the variable region with six bases added, or (4) the whole aptamer. Each of these selected classes showed an enrichment in TFBSs over unselected pools (with the exception of the whole aptamers in each of the Round 5 pools; Fig. 4). The selected pools all give the highest log likelihood score when considering the variable region plus six bases from the adjacent constant regions. This finding recapitulates what has long been observed with aptamer selections, especially aptamer selections that start with relatively short random sequence pools: aptamers that use fixed sequences to create high-affinity binding sites are at a numerical advantage relative to aptamers that use only random regions.
Conclusions
The automated selection protocol presented here successfully enriches aptamer pools for transcription factor binding sequences. Computational analyses show the selected pools contain more sequence matches to the set of known E. coli TFBSs than the unselected pool, and that the set of TFBSs is better represented in the selected sequences than is genomic DNA.
The fact that portions of the selected sequences could be fit to many different TFBSs contrasted with previous selections against complex mixtures, such as cell surfaces or human plasma. 16 In these instances, only a few aptamers predominated in the selections, and these aptamers were found to bind to only a few preferential targets. This highlights a key difference between selections for aptamers that bind transcription factors and selections for aptamers that bind to other proteins or targets, whereas all transcription factors and other DNA-binding proteins have a roughly equal capacity to bind relatively short DNA sequences, other proteins will have a widely variable affinity for nucleic acids.
Overall, our results suggest that it should be possible to characterize the protein-binding capacity of a lysate from virtually any organism by selection and computational analysis. It therefore may be possible to map previously unknown binding sites in newly characterized organisms in one step using aptamer sequences from selected pools. The automation of these procedures would correspondingly aid in mapping the transcriptome networks of entire sets of organisms.
Footnotes
Acknowledgments
LJS carried out liquid handling robot setup and programming, oligonucleotide design, cell transformation, lysate preparation, target plate setup, automated selection, sequencing, initial sequence analysis, and participated in manuscript drafting. PGS carried out scripting and sequence analysis, and participated in manuscript drafting. ADE acted in an advisory role, acquired funding, and participated in manuscript drafting.