
Abstract
An automated, high-throughput, open reading frame (ORF) library construction process has been developed. ORFs from genomic DNA of the microbe Sinorhizobium meliloti were amplified by PCR and cloned into the library vector by homologous recombination instead of traditional ligation. From 960 targets, we successfully generated 723 (75.3%) ORFs from the initial PCR. After cloning the successful samples into the library vector, transforming into E. coli and PCR colony screening, 371 (38.6% overall) ORFs were placed into the new library and sequenced. Our prototype library contained 314 (32.7% overall) clones with sequence identity to the Sinorhizobium meliloti genome.
Introduction and Background
Sinorhizobium meliloti is a well-studied, Gram-negative soil bacterium that stimulates the growth of root nodules in alfalfa and invades the nodules during their formation. There, the bacteria participate in a nitrogen fixation symbiosis with the plant. 1 S. meliloti has a relatively small genome, 6.6 Mb, which has been completely sequenced. The S. meliloti genome consists of a single chromosome and two large plasmids, which contain a total of ca. 6,200 predicted protein coding regions. 2 Since the genome is relatively small and has been sequenced, S. meliloti makes an ideal model organism for the development of an automated, high-throughput method for constructing an open reading frame (ORF) library. ORFs can be directly predicted from the genomic sequence from prokaryotes, which allows for rapid, automated primer design, and do not contain introns, which lends itself to the generation of a PCR-based library. Furthermore, this bacterium can be easily cultured under standard laboratory conditions, which allows for simple isolation of genomic DNA.
Creation of a library of S. meliloti ORFs will generate the necessary resources to facilitate analysis of S. meliloti DNA and proteins. Also, the techniques developed will expedite the production of libraries for other organisms. As the field of molecular biology moves toward automation, complete libraries will be critical for starting the process of automated genomic and proteomic analyses. Our group has expertise in working with genetic libraries such as the I.M.A.G.E. collection 3 and in automating high-throughput protein production, 4 which helped develop the techniques used to create the library.
The availability of whole genome sequences allows for rapid cloning of predicted ORFs and has begun to replace traditional cRNA library construction. Typically, pieces of genomic DNA are amplified by PCR and the purified products are cloned into vector DNA by ligation. The method described here replaces the inefficient process of ligation with a homologous recombination event (Figures 1). The In-Fusion™ system (Clontech; Palo Alto, CA) uses a proprietary enzyme to insert PCR products with the appropriate flanking sequences into a “donor” vector (Figures 2). Using the Creator system (Clontech; Palo Alto, CA), the cloned gene can be transferred from the donor vector into an “acceptor” vector applicable to a wide variety of expression systems. Similar recombination-based cloning systems are also available from other manufacturers; for example the TOPO Cloning kit allows for directional cloning of PCR products and is compatible with the Gateway system (Invitrogen; Carlsbad, CA). An important issue to consider when selecting a system is potential usage restrictions or licensing requirements; these vary by manufacturer.
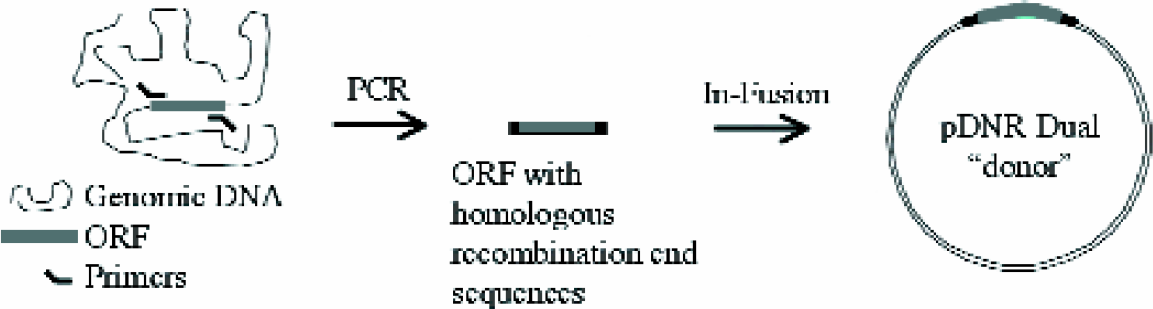
PCR primers, which incorporate the sequences needed for homologous recombination, amplify the ORFs from the genomic DNA. The PCR product is purified and inserted into the pDNR-Dual donor vector during the In-Fusion process by homologous recombination. The vector can be subsequently transformed into E. coli.
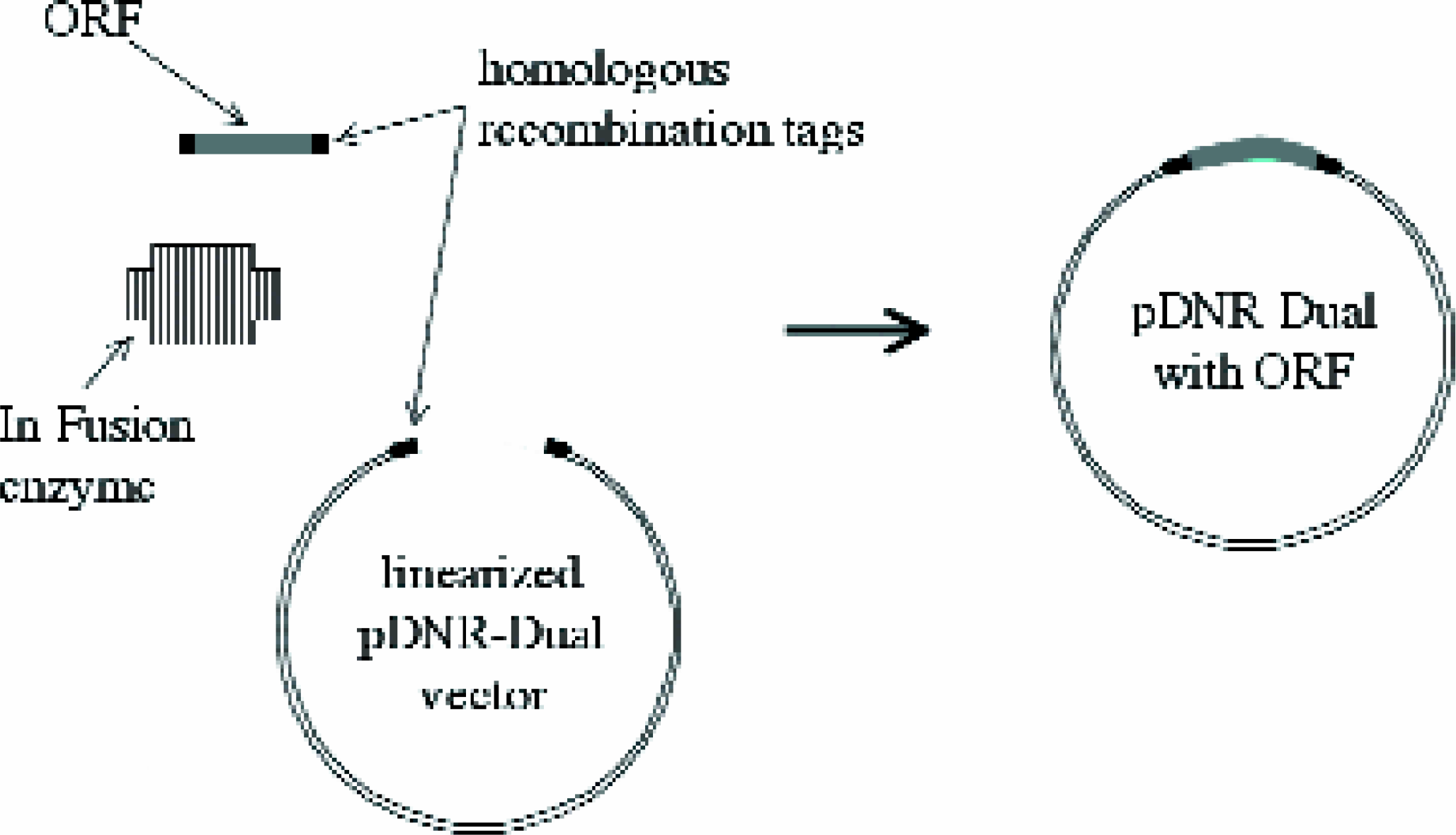
The In-Fusion enzyme facilitates homologous recombination between the purified ORF and the linearized pDNR-Dual donor vector. Since the vector contains loxP sites flanking the inserted ORF, the ORF can be easily transferred to a variety of acceptor vectors using Cre recombinase.
There are two important criteria for successful high-throughput molecular biology: the steps must be performed in the small volumes, e.g. in 96- or 384-well plates, and the steps must be automatable. If these two criteria are met, many samples can be processed in parallel and performed by laboratory robots. Parallel processing has a lower success rate than individual processing, since experimental conditions cannot be optimized for each sample. However, parallel processing expedites the entire process, therefore automating procedures increases the advantage of parallel processing and with optimization of automation, greater success can be achieved.
A thorough database is critical for tracking the large volumes of data generated by a high-throughput project. The database and its user interface should be designed for ease of use, and these should not only store information, but should also generate output to facilitate subsequent steps in the overall process. The database should apply the results to create input files for the robots and provide protocols and instructions for the user in order to minimize manual programming and the associated errors.
Creating DNA libraries is ideally suited for adaptation to high-throughput approaches, since most of the steps only involve straightforward liquid handling. Use of recombination-based cloning should increase the efficiency of the process, though it is a relatively new technology that will benefit from further optimization. S. meliloti is an ideal model organism, since it has a relatively small, fully-sequenced genome and an active research community eager to have the resources for its study.
Experimental Methods
PRIMER DESIGN
The primers were designed from the genomic sequence of S. meliloti in an automated process using a custom database program. The program copied the first and last 24 bases of the 5′ and 3′ end of each ORF and added a 16 base overhang for homologous recombination, as specified by the protocol for the In-Fusion kit, to create the forward and reverse primers. Primer statistics, including melting temperature (Tm), primer length, number of hairpins, and inter- and intra-primer annealing sites were calculated and stored in the database. Primers were clustered according to ORF size to facilitate downstream screening of the clones.
Primers were obtained at 200 μM concentration in TE buffer (20 mM Tris, 0.1 mM EDTA) with no purification (Illumina, Inc.; San Diego, CA). The primers were supplied in 96-well format for ease of automated processing. Primers were diluted to 5 μM in nuclease free water (Promega; Madison, WI) prior to use in PCR reactions.
INITIAL PCR
The initial round of PCR amplified the individual ORFs from S. meliloti genomic DNA such that the appropriate flanking sequences were added to the PCR products for insertion into the donor vector using the In-Fusion cloning kit. A PCR master mix (1x Reaction Buffer, 1 mM MgSO4, 2x PCRx Enhancer Solution, 1.25 units Platinum Pfx DNA Polymerase™ (Invitrogen; Carlsbad, CA), 0.3 mM dNTP mix, 10 ng genomic DNA template, and nuclease free water) was aliquoted into individual wells of a thin-walled 96-well plate using a custom program on a Genesis RSP 150™ liquid handling robot (Tecan U.S, Inc., Durham, NC). The robot was programmed to read an input file generated by the database interface to add the forward and reverse primers to the appropriate wells in a final concentration of 0.5 μM. The total reaction volume was 50 μl.
Platinum Pfx polymerase was used for the initial PCR since it provides rapid and high-fidelity amplification. The supplied enzyme is pre-inactivated by a bound antibody that is released during the initial denaturation step. Antibody inactivation is important in an automated reaction to avoid erroneous polymerization during the increased preparation time associated with 96-well plate PCR.
Thermocycling included an initial denaturation of 94°C for 2 min followed by 30 cycles of 94°C for 15 s, 50°C for 30 s, and 68°C for 3 min. All thermocycling was performed on a PerkinElmer Gene Amp PCR System 9600 Thermocycler (PerkinElmer Inc.; Wellesley, MA). Use of a hot lid thermocycler avoids the need for mineral oil, which can hamper automated sample handling.
To screen for successful amplification, 5 μl of each sample was analyzed on a 2% 96-well E-Gel. The E-Gel 96 Low Range DNA Marker was used as a standard (Invitrogen, Carlsbad, CA). After the gels were electrophoresed for 18 min, a digital image of the gel was captured and processed using the supplied software. The resulting amplified genes, as analyzed by the correctly sized bands in the processed images, were entered into the database.
To prepare the samples for cloning, PCR products were purified using a QIAquick 96 PCR Purification Kit (QIAGEN; Valencia, CA). Next, another Genesis RSP 150 custom program used a series of worklists generated by the database interface to rearray all of the positive samples into new 96-well plates.
CLONING BY IN-FUSION
The flanking sequences incorporated into each ORF in the initial PCR step allowed use of the In-Fusion PCR Cloning kit to clone each purified PCR product into the pDNR-Dual vector. Another custom program was employed on the Genesis RSP 150 to aliquot a master mix (1x In-Fusion Reaction Buffer, 50 μg/ml BSA, 5 ng/μl linearized pDNR-Dual vector, 20 units diluted In-Fusion Enzyme, and nuclease-free water) into each well of a thin walled, 96-well plate. The robot then transferred 5 μl of purified PCR product to the plate. The reactions were incubated at room temperature for 30 min.
Using a multi-channel pipettor, 1 μl of each In-Fusion reaction was transformed into TOP10 chemically competent cells in a 96-well plate (Multishot™ TOP10 96-well, Invitrogen; Carlsbad, CA). The cells were transformed according to the manufacturer's instructions and the entire reaction volume was plated on LB/agar plates containing 50 μg/ml ampicillin, 0.4 mM isopropyl-beta-D-thiogalactopyranoside (IPTG), and 40 μg/ml 5-bromo-4-chloro-3-indonyl-beta-D-galactopyanoside (X-gal) and grown at 37°C for 14–16 hours. Two white colonies from each plate were picked, since background colonies were blue resulting from incomplete linearization of the vector. Colonies were picked into 60 μl of storage media (sterile LB containing 8% glycerol and 200 μg/ml ampicillin) in 384-well plates, grown at 37°C for 14–16 hours, and then stored at −80°C.
COLONY SCREENING
To test for the expected In-Fusion cloning event, PCR colony screening was used. Custom programs directed the rearraying and dilution of overnight cultures on the Genesis RSP 150. A Multidrop™ (Thermo LabSystems Inc.; Beverly, MA) filled the dilution plates with sterile water, and a Hydra96™ (Robbins Scientific; Sunnyvale, CA) ensured that the samples were thoroughly mixed. The robots performed the 1:1000 dilution in two steps due to liquid handling volume limitations. In the first step, samples were rearrayed from a 384-well plate into a 96-well plate, in a 1:20 dilution. In the second step, the samples were diluted 1:50 in a new 96-well plate.
The PCR colony screening was performed in a similar manner to the initial PCR, with a few exceptions. The template DNA was derived from 10 μl of the diluted overnight culture rather than genomic DNA, and was added in a final step by the Genesis RSP 150. The necessary primer plates were identified by the database interface, which also generated a worklist file to control primer allocation by the robot. The primers used in the colony screening were the same as those used in the initial PCR. The master mix consisted of 1x Taq DNA Polymerase Buffer with MgCl2, 1.25 units Taq DNA Polymerase (Promega; Madison, WI), 0.3 mM dNTP, and nuclease free water. Thermocycling included an initial step of 95°C for 2 min, followed by 30 cycles of 94°C for 15 s, 50°C for 30 s, and 74°C for 3 min. Each sample (5 μl) was analyzed on a 2% E-Gel 96 for 12 min to screen for positives, and the results were entered into the database.
REARRAY/SEQUENCING/STORAGE
Following colony screening of ORFs from the 384-well plates, the database interface generated a Genesis RSP 150 worklist for rearraying the positive samples into new 96-well plates containing storage media. The plates were incubated for 14–16 hours at 37°C and then stored at −80°C. The 96-well storage plates were replicated and subsequently sequenced.
DATABASE DESIGN
The custom Oracle 9i database, residing on a Solaris 8 system, consists of 27 tables (Figures 3) that can be grouped into four sets:
ORF data, including primers plate and well data protocols process data, including results.
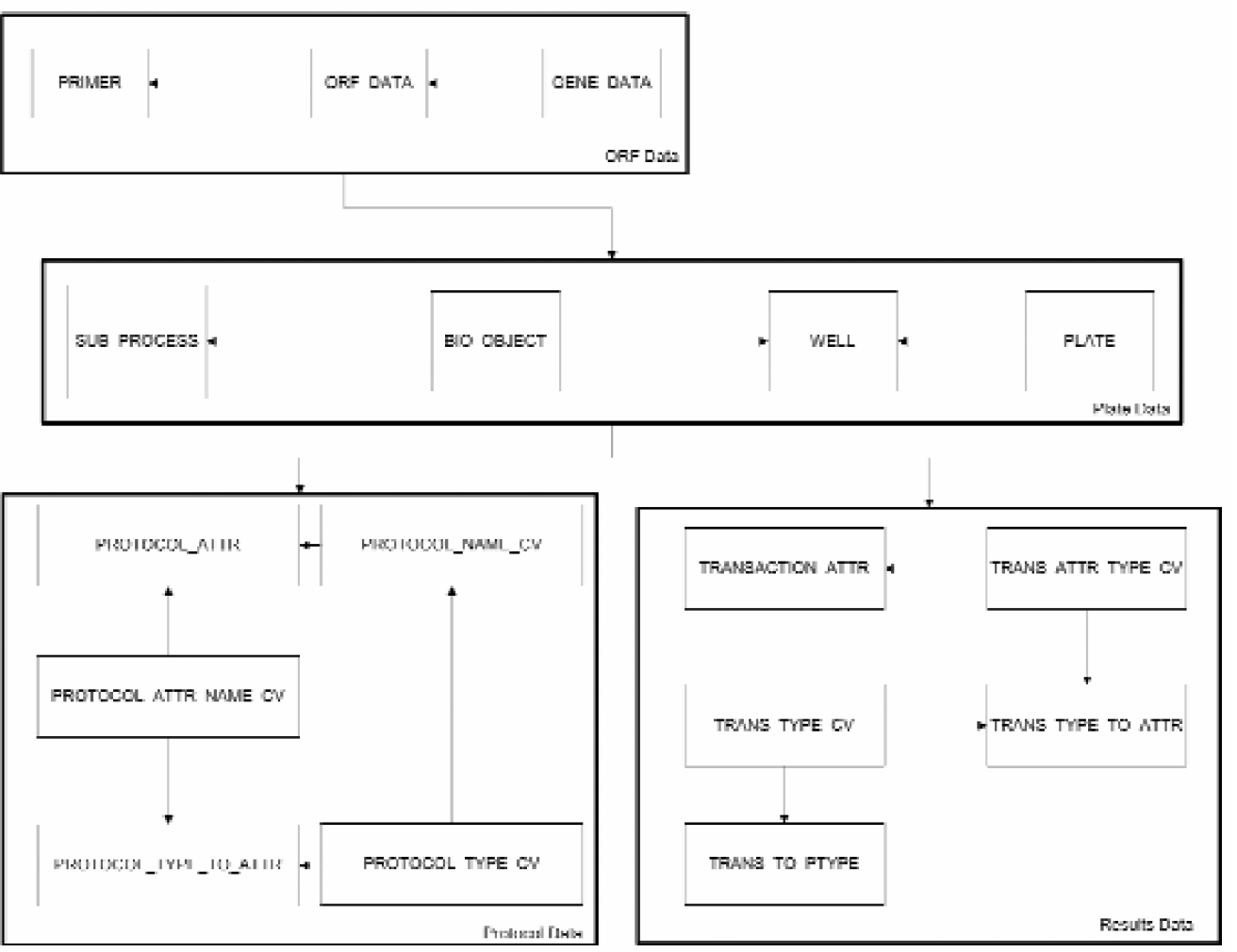
Database design for the cloning process is based on four main data types: 1) ORF, gene and primer information, 2) Plate locations, 3) Protocols and 4) Results. These tables are linked to efficiently allow the clones to be tracked throughout the process.
The data is tracked by the unique ORF, each of which has been assigned a primary key. Given this key, users have the ability to access or update the status and results of any ORF in the pipeline.
1. ORF data
The vector table contains the name, source, and antibiotic resistance of the vector that was used with each primer. The gene_data table contains information about each gene in the database - name, function and comments and is linked to the orf_data table, which contains the name, coding start and stop, sequence length, GC content, accession number, species, strain and sequence of each ORF.
The primer table contains the primer status (successful, unsuccessful, untested), name, endedness, sequence, linker, melting temperature, sequence length, and number of annealing sites. It is also linked to primer_status_cv, a controlled vocabulary table for the primer status field, which restricts the selection of input choices. Key table linkages enable effective data mining, these include linking gene_data, bio_object, orf_data, and vector information.
2. The plate and well data
The plate table contains plate status, type, name, number, version, barcode, and freezer number. It is linked to the well table, which lists the row and column position of each well on the plate, and links to the bio_object table to track contents. The bio_object table tracks the contents of each well, the status of the well in the process, and the type of the object (primer, amplified ORF, ORF, etc.). The bio_object table is linked to the sub_process table, which tracks the history of each ORF. The sub_process table tracks the ORFs or primers that were used as input to the process and the ORF object that was produced. The protocol and results can be stored and obtained from the sub_process table.
3. Protocol data
The protocol_type_cv tracks all types of protocols, e.g. PCR protocol, transformation protocol, etc., and the fields to be stored are defined based on that type. This table links to protocol_type_to_attr, to define those fields. This table stores the attribute names, and links to protocol_attr_name_cv, which defines the values of attribute names. Next in the process is protocol_attr, which stores the protocol data, and then protocol_name_cv, which stores the name of each defined protocol.
4. The process and result data
One table, trans_to_ptype defines the input and output plate type for each process. The remaining tables are very similar to that for the protocol tables. The first of these, trans_type_cv, defines the process types - create PCR plate, dilute plate, etc. It links to action_results, which helps to define when results should be expected. It also links to trans_type_to_attr, which defines the fields in which to store results for that plate type. This links to trans_attr_type_cv, which defines the possible names for each field. Lastly, this is linked to transaction_attr, which stores the actual results and data.
Results and Discussion
OPTIMIZATION OF LIBRARY CONSTRUCTION
The ORFs from S. meliloti vary in size from approximately 100 to 8500 bp with an average size of 930 bp, and so the primers were designed with a range of melting temperatures from 45°C to 71°C. Choosing thermocycling temperatures was further complicated by the 62.1% GC content 2 of the genomic DNA. As such, the Platinum Pfx DNA Polymerase system, which included the PCRx Enhancer solution, was employed to attempt to alleviate these problems. PCR was performed using 0.5x, 1x, 2x, and 3x concentrations of the PCRx Enhancer solution. Based on initial testing, all subsequent samples used a 2x final concentration of the PCRx Enhancer solution (data not shown).
Purified PCR products were rearrayed and the In-Fusion reaction was performed. Transformation of the In-Fusion reactions was tested with several different cell lines. Initially, DH10B-T1 phage resistant cells (Invitrogen; Carlsbad, CA), which are advantageous for library construction since they provide a high efficiency of transformation as well as affording protection against T1 bacteriophage, were used. However, the cells were not commercially available in 96-well format, and the competent cells made in-house did not efficiently take up the subcloned ORFs. Fusion-Blue cells (Clontech Laboratories, Inc.; Palo Alto, CA) were also tested and worked efficiently; however, they were not commercially available in 96-well or bulk format. Finally, TOP10 chemically competent cells (Invitrogen, Carlsbad, CA), available in a 96-well format, were transformed and were as efficient as the Fusion-Blue cells. Plating the entire transformation reaction (200 μl) produced approximately 100–200 colonies per plate.
PRODUCTION OF ORF LIBRARY FROM S. MELILOTI
For all 960 primer pairs, the average Tm was 58°C ± 4°C, the average number of intra-primer annealing sites was 4.2 ± 2.7, and the average number of inter-primer annealing sites was 3.0 ± 2.0. After the initial PCR, 723 ORFs (75.3%) showed the expected band at the appropriate size after gel electrophoresis. The primers for the positive samples had an average Tm of 57°C ± 4°C, the average number of self annealing sites was 3.7 ± 2.6, and the average number of inter-primer annealing sites was 3.2 ± 2.0. There was no pattern to these primer characteristics for the ORFs that were unsuccessful, nor was the size of the ORF indicative of success or failure of the amplification. For subsequent attempts at PCR, the ORFs will be clustered by primer Tm, and PCR protocols will be employed that vary temperature in order to increase the efficiency of the PCR.
IN-FUSION REACTION, TRANSFORMATION, AND COLONY SCREENING
The 723 positive ORFs were cloned by In-Fusion, transformed into TOP10 chemically competent cells, and the entire transformation volume was plated on LB/agar plates containing ampicillin, X-gal, and IPTG. The results showed that 89% of the agar plates had at least 2 colonies, 94% had 1 or more colonies, and only 6% had no colonies. The colonies were picked into liquid culture in a 384-well plate and grown for 14–16 hours. One set of 96 failed to grow due to probable contamination of the media used to make agar plates.
Subsequently, the colonies were screened by PCR and the expected insert was identified in 371 ORFs. The efficiency of the In-Fusion/Transformation/PCR colony screening process was 51% (371/723). One potential limitation to In-Fusion cloning is that the In-Fusion reaction is potentially toxic to the cells, 5 so only 1 μl of the reaction could be transformed. The manufacturer is currently investigating ways to increase the amount that can be transformed by neutralizing the toxicity of the reaction as well as optimizing protocols for diluting the reaction to allow for transformation of a higher volume. In the future, we will employ these new methods, which should increase the number of positive clones obtained. However, even without increasing the amount of reaction transformed, retrying the negative samples would likely prove worthwhile for increasing the total number of positive clones.
Currently the transformation, plating, and picking processes are not automated. Use of specialized robots that could automate these processes would dramatically increase the throughput and efficiency of the system. The colony picking robots available did not have software immediately amenable to this process; however, in future work, software will be developed that will allow further automation of the overall process.
SEQUENCING
Of the 371 samples that were 5′ end sequenced, 314 (32.7% overall) showed that the expected ORF sequence was correctly cloned into the pDNR-Dual vector. Of the samples that did not correctly verify, the majority were due to low quality sequence data (48 of 57). Of the remaining nine high quality samples, eight represented cloned ORFs present in the incorrect well location (duplicate clones), and the remaining sample did not match anything in Genbank. Since ORF isolation from genomic DNA was performed via PCR with a proofreading polymerase, and the sizes of the inserts were checked against the expected ORF sizes, 5′ end sequencing was sufficient to determine that each sample was successful. In future projects, especially those where the downstream application is dependent on error-free amplification such as protein structure determination, full sequence verification of the entire insert could be performed if necessary.
DATABASE AND USER INTERFACE
The database was initially populated with data for all 6200 ORFs predicted from S. meliloti, obtained from NCBI, from which 960 clones were analyzed. Next, plate and well data about the primers that were designed for the subset of ORFs that were tested were entered using tab-delimited spreadsheets supplied by Illumina. Plates for subsequent steps of the process were generated, named and entered automatically for the user and were based on stored data and, in most cases, some user feedback. At several stages, when the plate was designed, the interface also provided a worklist file for the Genesis RSP 150 and a text file for the user, providing instructions regarding construction of a new plate. The text file contained a list of the plates required and a list of the ORF names and positions on the new plate. By this method, the ORFs were tracked automatically by the database throughout the process.
The database was designed to support a graphical interface with fields that could accept all the different types of data generated in the process. Initially, each field required manual input of data. To streamline the process, the ability to input results via a tab-delimited spreadsheet file was added. The graphical interface is available for the addition of comments and the modification of data. Finally, storing all of the data in this fashion allowed for the creation of a summary webpage that displays the current results, including number of successes and failures, efficiency of each step in the process, and the location of each ORF in the plates.
Overall Results
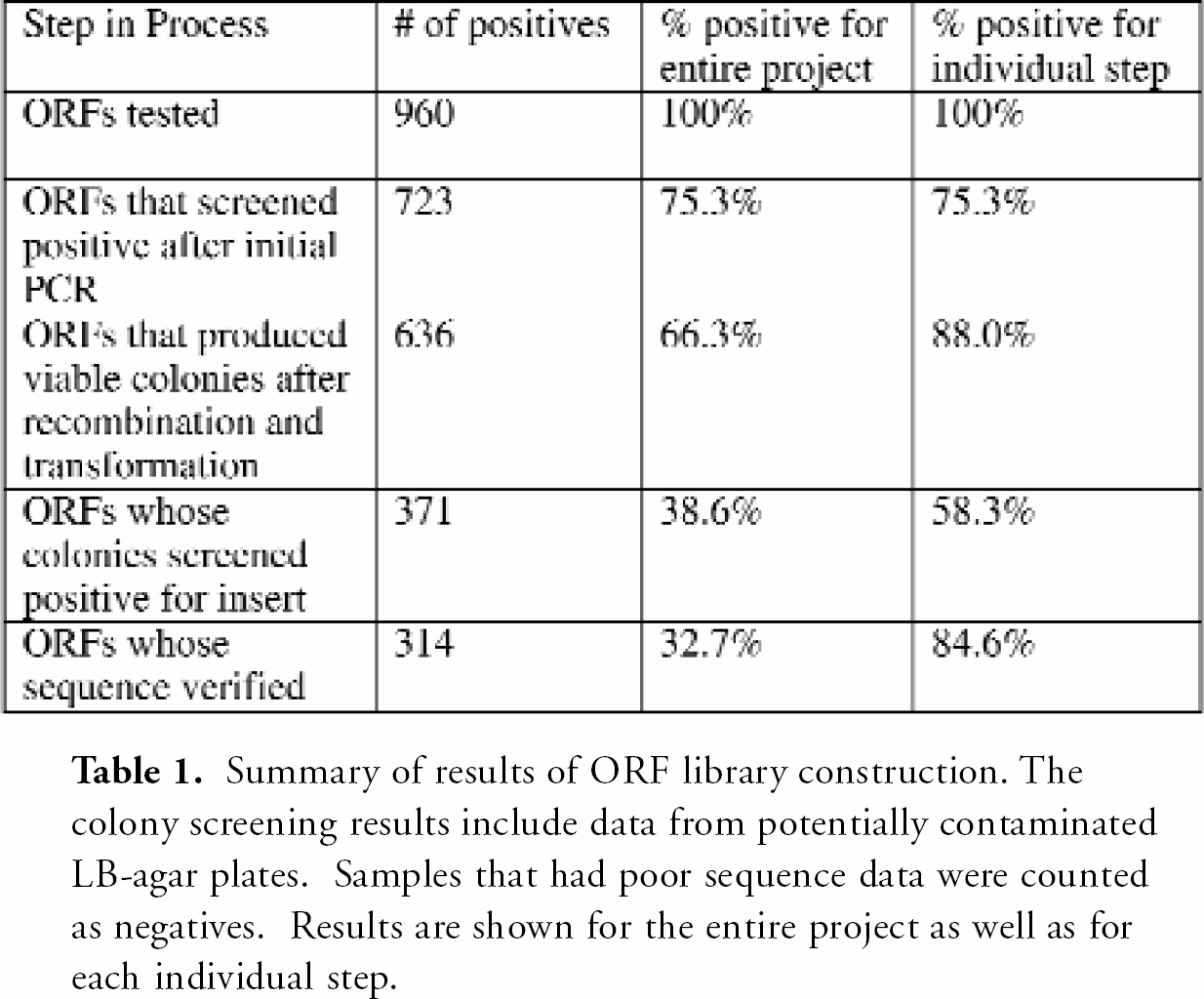
The results of our prototype library construction are summarized in Table 1 Disregarding the data from the 192 ORF clones affected by the contaminated reagents described above, the final results show that 39.9% overall (314/786) were successfully cloned by the In-Fusion method and sequence-verified.
Summary of results of ORF library construction. The colony screening results include data from potentially contaminated LB-agar plates. Samples that had poor sequence data were counted as negatives. Results are shown for the entire project as well as for each individual step
Each ORF was only processed once. In future production, each ORF that fails at a given step will be repeated, thus increasing the overall efficiency of the final library construction process. Additionally, PCR conditions could be further customized for samples that failed the initial round of PCR by altering the conditions to fit the given samples on each plate. Finally, future experiments will take advantage of refined In-Fusion protocols that aim to provide even greater efficiency in ligation-free cloning.
Conclusion
We have successfully prototyped an automated process for generating a bacterial open reading frame library from purified genomic DNA. The process requires sequence information for primer design to amplify the ORFs of interest from genomic DNA, and libraries for other organisms of interest could easily be constructed for those additional organisms whose genomic sequence is available. Moreover, a large cDNA collection is available from both human and mouse genome efforts which could also serve as templates for expression libraries using this system. 6 Cloning was performed by homologous recombination instead of ligation in order to increase efficiency as well as to facilitate automation of the process. The ORFs were cloned into a donor vector that is compatible with the Creator system from Clontech, so they can be easily transferred to multiple expression systems, such as those used for high-throughput protein production. 7 Most of the steps in the process are currently automated, and the remaining steps were designed such that they can be automated in the future without redesigning the process.
Acknowledgments
The authors would like to thank Dr. Turlough Finan for his scientific expertise in Sinorhizobium meliloti and for his generous contribution of genomic DNA, and Dr. Andrew Farmer for his assistance in optimizing the In-Fusion cloning system. This research was supported by a LLNL-Lab Directed Research Directorate award to C. K. P. This work was performed under the auspices of the U.S. Department of Energy by the University of California, Lawrence Livermore National Laboratory under contract no. W-7405-ENG-48.