
Abstract
Locked Nucleic Acid's or LNA are a new class of bicyclic DNA analogues that have a high affinity and specificity towards complementary nucleic acids. LNA containing oligonucleotides were used to develop a multiplex SNP genotyping assay based entirely on hybridization between capture probe and target. The approach incorporates a polymer microarray platform, photochemistry for immobilization of oligonucleotides onto microarrays, and a dedicated software tool to aid primer and capture probe design for highly multiplex genotyping. Furthermore, these technologies are combined in an integrated microfluidics platform for simple, highly multiplex and robust SNP genotyping.
INTRODUCTION
Single nucleotide polymorphisms (SNPs) are the most common form of human genetic variation as SNPs occur at a frequency of 1/1000 base pair in the human genome. 1 Understanding these complex patterns of genetic variation holds promise in many aspects of disease control: i) as diagnostic markers for monogenetic diseases as well as for more complex genetic disorders, 2 3 in the field of theranostics where individual drug responses may be correlated to a specific SNP-profile, 4 and iii) as prognostic tools to predict the risk of a particular disease. 5
The high demand for SNP genotyping requires robust high throughput, multiplex and cost effective SNP genotyping. In most cases, DNA-DNA hybridization assays are not capable of discriminating single mismatched base pairs, which have forced development of various technologies using enzymes in order to obtain the needed specificity for SNP identification. Such enzyme dependent assays are for instance: primer extension assays, 6 7 oligonucleotide ligase assay 8 and the Invader assay. 9 Whitcombe et al., 10 demonstrated the use of TaqMan probes for homogeneous detection of SNPs. In addition, allele specific hybridizations with probes designed as molecular beacons 11 have proven useful. However, none of the homogenous assays are easy to multiplex to a level comparable to microarray based systems.
Here we present a SNP genotyping technology based on the DNA derivative LNA (Locked Nucleic Acid) 12 which offers the needed specificity for SNP genotyping by hybridization without the need for enzymes. The approach consists of five key technologies: 1) LNA, which allows multiplex target amplification and SNP genotyping by hybridization, 2) a polymer microarray platform for superior spot quality, 3) anthraquinone photochemistry for covalent immobilization of macromolecules to the polymer platform, 4) a software tool for efficient design of microarray capture probes and primers for multiplex PCR amplification of target DNA, and 5) a microfluidics microarray platform for low volume SNP genotyping. The present paper describes how these technologies are combined into a simple and reliable SNP genotyping system.
MATERIALS AND METHODS
Design and synthesis of PCR primers and capture probes
Primers and capture probes were designed using the novel software EURAYdesign (Ver.1.0), which is capable of handling large sets of SNP data. This software package automatically optimizes the position of capture probes for SNP detection according to a number of parameters like cross hybridization, Tm, GC-content, self complementarity, etc. EURAYdesign has incorporated algorithms for PCR primer design from Primer 3, 13 and an additional number of algorithms useful for the design of primers for multiplex PCR amplification and SNP detection.
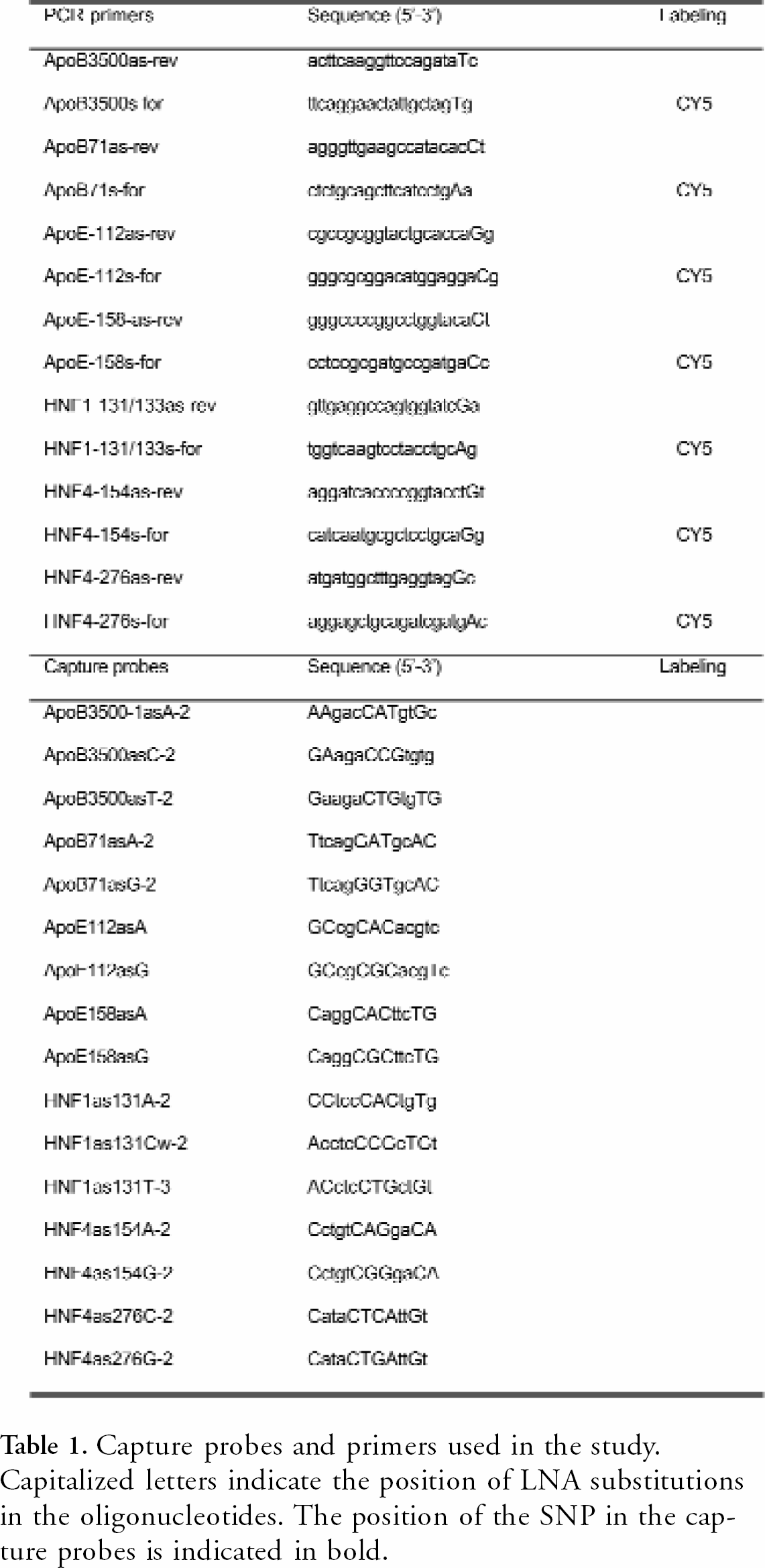
All probes and primers containing LNA (Table 1) and anthraquinone were synthesized using standard phosphoamidite chemistry on an Expedite DNA synthesis machine (Model 8909 equipped with Multiple Oligonucleotide Synthesis System (MOSS) Applied Biosystems, Framingham MA), purified by HPLC and analyzed by MALDI-TOF mass spectrometry. Capture probes were synthesized with a linker with an anthraquinone in the 5' end. The PCR primers and capture probes used in the study are shown in Table 1. 30mer synthetic targets used to validate the capture probes were DNA oligonucleotides containing 15 bp of the target sequence on each side of the SNP, and were synthesized by DNA Technology, Aarhus, Denmark.
PCR
The PCR primers were designed as described above with the forward primers being CY5-conjugated. For multiplex detection of the nine SNPs, the seven PCR primer pairs were pooled into one multiplex PCR, carried out using 100 pg/μl genomic DNA (Promega DNA G304A lot# 9273101, Promega WI), 400 nM of each forward and reverse primer, 0.2 mM dNTPs, 3 mM MgCl2 and 5 U AmpliTaq Gold DNA polymerase (Applied Biosystems, CA) in 200 μl GeneAmp Buffer II supplied with the enzyme. The final volume was 200 μL. The enzyme was initially activated at 95°C for 5 minutes, then 35 cycles of the following parameters were run: 94°C for 45 seconds, 56.6°C for 45 seconds and 68°C for 1 minute, followed by a final elongation for 10 minutes at 60°C. The singleplex PCR reactions were run using the same conditions as described above for multiplex PCR.
Capture probes and primers used in the study. Capitalized letters indicate the position of LNA substitutions in the oligonucleotides. The position of the SNP in the capture probes is indicated in bold.

Spotting
10 μM solutions of the 18 capture probes, in 100mM phosphate buffer (pH = 7.2), were spotted onto EURAY polymer slides (Exiqon, Vedbaek, Denmark) using a BioChip Arrayer I (Packard BioChip Technologies, Meriden, CT). Alignment spots consisting of a linker modified with an anthraquinone at the 5′ end and a biotin in the 3′ end were included in the array. Five replicates of a 4×11 array were spotted on the EURAY polymer slide. Each array contains a row of four markers at the top and at the bottom, to indicate the outer boundaries of the array. The capture probes were covalently attached as described by Koch et al., 12 to the polymer slide by UV light irradiation for 2500μl in a Stratalinker 2400 (Stratagene, La Jolla, CA). Subsequently, the slides were washed in highly purified water for 2 hours to remove salts and excess of capture probe. The slides were finally dried for 30 min. at 37°C in an oven.
Hybridization
For hybridization with synthetic biotin-conjugated 30mers the oligonucleotides were diluted to a concentration of 0.01μM in 1×SSCT (1×SSC+0.1% Tween; 20×SSC is 3.0M NaCl, 0.3M sodium citrate, pH 7.0) denatured for 5 min at 95 °C, and hybridized to the spotted slides for 2 hrs at 37°C. For multiplex hybridization, the synthetic biotin-conjugated 30mers were pooled, and diluted to a final concentration at 0.001 μM in 1×SSCT, denatured for 5 min at 95 °C prior to hybridization to the spotted slides. The hybridization area was covered with a coverslip (Grace coverslips, Sigma St. Louis, MO) and conducted overnight in a moisture chamber at 37°C. Slides were subjected to a two hour stringent wash at 37°C in 0.15×SSCT For detection the slides were incubated using a 0.33μg/ml solution of CY5-conjugated streptavidin (DAKO A/S, Denmark) in 1×SSCT for 30 minutes at 37°C, washed one hour at 37°C and finally air-dried for 20 minutes at 37°C.
The PCR amplicons directly labeled with CY5 were purified and concentrated using QIAquick spin columns (QIAGEN, Germany). 17.5×SSCT+ 50% dextran was added to the eluate to a final concentration of 3.5×SSCT+10% dextran (Sigma, St. Louis, MO). The amplicons were denatured for 5 min at 95 °C, and hybridized to the spotted slides overnight at 37°C, followed by two hours wash at 37°C in 1×SSCT, and finally air-dried for 20 minutes at 37°C.
In the integrated microfluidics platform multiplex hybridization was performed using pooled synthetic biotin-conjugated oligonucleotides diluted to a final concentration of 0.01μM in 4×SSCT 70μL of the target solution was added, and the chip was hybridized over night at room temperature. The target solution was then flushed out of the analysis chamber by applying 200μL of air through the sample port with a standard pipette. For detection, 70μL of a solution of CY5-labelled streptavidin (DAKO A/S, Denmark) at 1 μg/mL in 5×SSCT was added and incubated for one hour. The chip was emptied with 200μL air, and subsequently washed with 200 μL of 0.15×SSCT. The chip was incubated with 0.15×SSCT for one hour, emptied with 200 μL air, and re-filled with 0.15×SSCT.
Slide scanning and image analysis
Two polymer platforms were used. An open EURAY slide system (Exiqon A/S, Denmark) with dimensions similar to a glass slide, and the EURAY SNP-chip with integrated microfluidics.
Slides were scanned using an arrayWoRx scanner (Applied Precision, Issaquah, WA) equipped with standard filter sets for Cy5 fluorescence detection. Spot intensity was quantified by image analysis using Array Vision (Imaging Research, St. Catharines, Ontario, Canada). For each spot, the mean value was calculated, and local background subtracted.
Images of hybridizations conducted in integrated microfluidics platform was acquired using a Zeiss fluorescent microscope equipped with an XBO lamp, an emission/excitation filter set of 650nm/670nm and a 5× objective. Images were captured using a Coolsnap camera (Photometrics, Tucson, AZ) using 5000msec integration time. Data analysis was conducted as above.
RESULTS
For cost effective SNP genotyping a high degree of multiplexing of both target amplification and genotype determination is required. Here we demonstrate the application of a polymer slide platform as well as integrated microfluidics chip for multiplex genotyping of 9 single nucleotide polymorphisms, based on just one multiplex PCR reaction for target amplification. The SNPs included on the array are related to Alzheimer's (ApoE), coronary heart disease (ApoB), and diabetes (HNF1 and HNF4), and serve to demonstrate the ability of genotyping different disease related genes.
Capture probe design and validation
For optimal capture probe and primer design in highly multiplex systems, parameters like non-specific cross hybridization, self-dimer formation and Tm of capture probes and primers must be taken into account. Commercial primer design-programs are available, but they are not developed to accommodate simultaneous PCR primer and capture probe design in highly multiplex situations, where factors like cross-hybridization become very complex. For that purpose, we have developed a dedicated software tool, termed EURAYdesign, which takes all the above-mentioned parameters into account. In an iterative process, the program will identify the optimal PCR primers resulting in the lowest degree of cross hybridization between amplicon and capture probe as well as primer pairs that are optimal for multiplex PCR. Furthermore, EURAYdesign incorporates features for optimizing the position of LNA nucleotides in both capture probes and PCR primer sets, and this is not possible using commercial primer-design programs.
Due to the high affinity and specificity of LNA towards complementary DNA, and its compatibility with standard DNA synthesis, LNA can be mixed with DNA in order to create shorter and more genotype specific capture probes. In general, we use capture probes consisting of 12 nucleotides mixed with 3–5 LNA nucleotides, giving the needed specificity and affinity for SNP detection. Figure 1 shows the application of EURAYdesign to design capture probes and multiplex PCR primer sets. The set of genes are shown graphically depicting the position of the SNPs, and the position of the capture probes and primers.

Primer and capture probe design using EURAYdesign. Grey lines represent the sequence surrounding the SNP, which is marked with black vertical lines. Black regions are possible primers and grey regions are optimal primer in the present context. The position of the capture probe is indicated by the dark grey line covering the SNP. The toggle in the lower left corner optimizes the position of the LNA nucleotides in the capture probe online with an estimate of the Tm value.
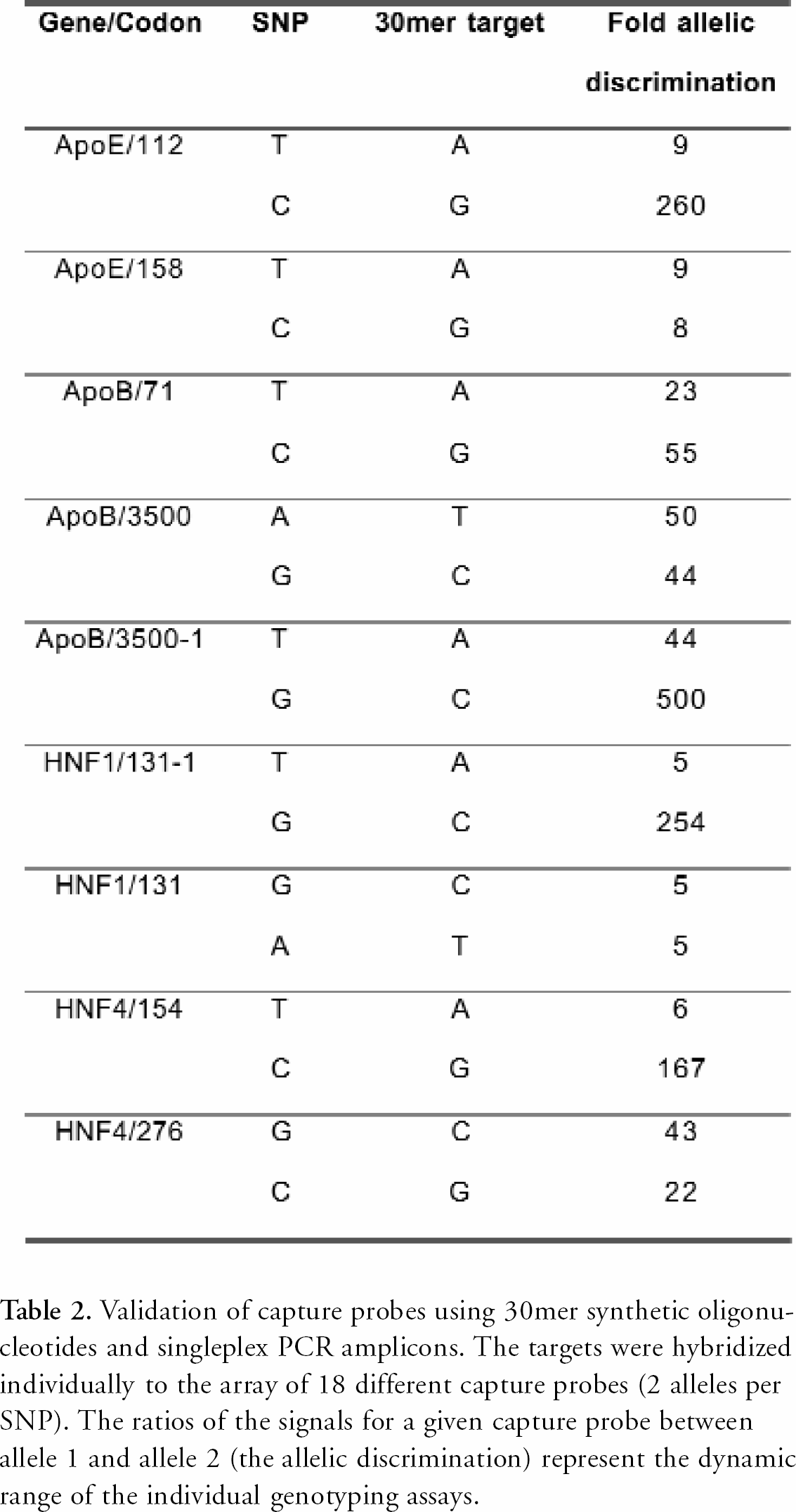
The oligonucleotides suggested by EURAYdesign were synthesized off chip, quality checked and spotted onto the EURAY polymer slides. The specificity of each SNP capture probe pair was validated using a synthetic 30mer DNA target, by hybridizing each target individually to the array. The signal-to-noise ratio of the genotyping assay was defined as signal from the perfect match (allele 1) divided by signal (noise) from the single mismatch (allele 2) situation. The results presented in Table 2 demonstrate that chimeric capture probes of LNA and DNA enable single nucleotide genotyping by hybridization with a significant allelic discrimination.

Verification of the 7-plex PCR reaction. Lanes 1 to 7 show the individual PCR reactions. The sizes are lane 1: 50bp, lane 2: 56bp, lane 3: 63bp, lane 4: 111bp, lane 5: 68bp, lane 6: 90bp and lane 7: 49bp. Lane 8 shows the combined multiplex PCR reaction where the 50/49 bp band is a double band. Lane 9 is a 100 bp DNA ladder.
Validation of capture probes using 30mer synthetic oligonucleotides and singleplex PCR amplicons. The targets were hybridized individually to the array of 18 different capture probes (2 alleles per SNP). The ratios of the signals for a given capture probe between allele 1 and allele 2 (the allelic discrimination) represent the dynamic range of the individual genotyping assays.
In addition, singleplex PCR amplicons generated from pooled genomic DNA from 50 American individuals were hybridized individually to the array in order to validate that the flanking regions included in the amplicons did not cross-hybridize. All singleplex amplicons hybridized specifically to one or both of the appropriate alleles (data not shown). The allelic specificity was not determined as the actual genotype of the genomic DNA mixture was not known.
Multiplex target amplification
The high binding affinity of LNA enables genotyping of a given set of SNPs using one multiplex PCR reaction and a single microarray. The 9 SNPs genotyped on this array were amplified using a single 7-plex PCR reaction, since two of the amplicons contained two SNPs each. All PCR primers contained a single LNA nucleotide one nucleotide from the 3' end of the primer. LNA at this position significantly enhances the specificity of primer pairs and enhances the ability to perform multiplex reactions. Figure 2 shows a gel verifying the presence of all amplicons in the multiplex PCR amplification. The 49/50 bp band is a double band, and both amplicons were present as evidenced by hybridization (data not shown) since we know that the amplicons do not cross-hybridize with unrelated capture probes.
Genotyping of genomic DNA
Figure 3 shows genotyping of genomic DNA using the 9 SNP array. A single multiplex PCR reaction on the genomic DNA mixture from 50 individuals was performed, and amplicons were purified and hybridized to the array. The prevalent genotype in the genomic DNA mixture is marked in bold types in Figure 3. Notice that the image demonstrates genotyping of DNA pooled from 50 individuals thereby lowering the signal to noise ratio as the pooled DNA is likely to be heterogeneous.

Genotyping of nine SNPs using the EURAY platform. The microarray was hybridized with amplicons from the multiplex PCR reaction, and the image demonstrates the resulting genotyping. The table designates the sequence of the allele positioned on the array. All capture probes were spotted in duplicate. The dominant genotype of this mixed DNA pool is highlighted in bold.
Application of the EURAY SNP-chip for integrated genotyping
For applicability in large genotyping studies SNP analysis has to be performed in parallel and in a highly reproducible fashion. Automated hybridization stations are one solution to this problem. To ease handling and increase the reproducibility of genotyping assays we have developed an integrated microfluidics platform – the EURAY SNP-chip (Figure 4A) – that allows all post amplification assay steps to be integrated on the same device. The device incorporates a liquid inlet port designed to interface with a standard pipette tip, a hybridization chamber shaped as a meandering channel, to obtain a large analysis area and a hydrophobic stop to prevent the liquid from being drawn out of the analysis chamber by capillary forces. The device further comprises an integrated waste chamber capable of holding all sample and buffers used during the assay. The chip requires loading with as little as 45 μL of target DNA solution, which is considerably less than most hybridization stations. The array was spotted on the inside of the lid. The lid was then placed on the base structure containing the microstructures and infrared (IR) transmission laser welding was used to seal the chip. The EURAY SNP-chip was hybridized with a mixture of synthetic 30mer targets. Figure 4B shows the genotyping conducted in the microstructure.

Genotyping using the EURAY SNP-chip. A The microfluidics structure with inlet, meandering hybridization chamber and the exit to waste chamber. A waste reservoir is placed on the opposite side of the chip (not seen). B The genotyping using the integrated microfluidics platform. For ApoE112, ApoE158, ApoB71, ApoB3500–1, HNF1–131, HNF1–131–1, HNF4–276 allele 1 was added, for ApoB3500 and HNF4–154 allele 2 was added to the microarray. A pseudocolor scale is included for reference.
An inherent feature of hybridizations on microarrays is that different capture probes hybridize with different affinities. This implies that the signal from one perfect match hybridization can be in the same range as the mismatched signal from another set of capture probes (Fig 4B compare ApoE112 with HNF1–131). However, this does not matter as the capture probes are unrelated and the amplicons do not cross-hybridize. Including LNA in the capture probes facilitates design of very specific and non-crosshybridizing capture probes. In the case of the ApoE112 and HNF-131, the genotype calling was unambiguous as the signal to noise between the two alleles of each SNP was still significant.
In conclusion, we have demonstrated a new approach to SNP genotyping that based on LNA technology offers enzyme independent specificity in genotyping by hybridization. We have described two new polymer microarray platforms, an open slide format and a closed system with integrated microfluidics. Capture probes are immobilized covalently to the polymer surface by simple photoactivated chemistry facilitating homogeneous spots with even distribution of the capture probes within the spots. Furthermore, we have demonstrated a new software package, which facilitates in silico design of microarrays with optimized capture probes and PCR primers for multiplex target amplification.
The technology not only allows simple, accurate and cost-effective genotyping as described in this paper, but also very sensitive expression analysis using microarrays.
ACKNOWLEDGEMENT
The polymer platforms were developed in collaboration with STEAG microParts, Germany. We would like to thank Mette Bjørn, Dana Tvermoes, Annemette Borch, Mette Randahl Larsen and Morten Bruus for technical assistance.