
Abstract
The analysis of biological screening results has traditionally been a labor intensive process. Scientists familiar with the biological data under investigation would visually inspect the results, evaluate the quality and promise of active data points and identify leads. The introduction and widespread use of high throughput screening systems has increased the size of biological datasets immensely thus pushing the traditional analysis method to its limits. In this presentation we describe a new automated approach that emulates the decision making process of human experts. This approach combines knowledge-based techniques with human expertise to enable rapid identification, characterization and prioritization of lead candidates.
AUTOMATION AND BOTTLENECKS
The necessity for the introduction and wide use of automation in the modern drug discovery process is well-known and accepted. 1 The overwhelming complexity of the drug discovery process, the increased complication of both targets and corporate libraries and the higher market demands require advancements in the process to make it more efficient and effective. In this respect, automation could be particularly useful in eliminating current bottlenecks, improving the quality of the results, reducing the time and cost of the process and thus, increasing the throughput of the process.
In the past decade the pharmaceutical industry placed emphasis on automating the screening and synthesis of compounds. As a result, High-Throughput Screening (HTS) and Combinatorial Chemistry are currently the norm rather than the exception in the process. However, automation of these two steps of the drug discovery process resulted in the creation of new bottlenecks further downstream. As a consequence the overall throughput increase of the drug discovery process is not nearly as much as expected. The investment in money, time and effort has not yet resulted in an increased number of drugs discovered.
AUTOMATION IN DATA ANALYSIS
Automating the analysis of screening data produced by HTS systems is a formidable task. HTS datasets are comprised of large amounts of complex and noisy data. This type of data poses a serious challenge to most of the widely used computational algorithms and tools. In addition, the analysis needs to be of high quality and accuracy due to the importance of the decisions based on its results. Wrong decisions at this crucial step can lead to a tremendous waste of resources, increased costs, and a significantly longer time for the completion of the drug discovery cycle.
CURRENT DATA ANALYSIS METHODOLOGY CHARACTERISTICS
The current process of screening data analysis is human intensive. Human experts, usually medicinal chemists, have to visually inspect the data after limited computational processing and reach conclusions based on their expertise and knowledge of the assay. This dependency on humans limits the throughput of the process. Nevertheless, before the introduction of automation in the form of HTS systems those limits were a non-issue since the amount of screening data allowed the process to be effective. With the relatively recent widespread employment of HTS and ultra HTS (uHTS) systems the data overflow problem has surfaced as a major bottleneck impeding scientists from accessing and using all the knowledge the screening process generates.
As noted above, limited computational processing of the screening dataset is commonly performed in order to simplify the task of the expert. This sort of computational processing is mainly aimed at reducing the size of the dataset that the expert needs to consider to manageable levels. Various techniques including modeling tools, visualization packages, similarity metrics, substructure search methods and clustering and classification methods are widely used to this end. A typical example is sorting or clustering the screening results based on measured activity and bit-string based representations of chemical structure and placing activity thresholds to eliminate the majority of compounds as inactive. 2,3,4,5
This type of computational processing, although useful, readily available and able to meet the data reduction goals set, has serious inherent limitations. Most importantly, it ignores all information that can be found from compounds showing low or no activity and neglects the presence of noise in the data that often takes the form of false positives and negatives. In addition, a user must be an expert in the technology and a number of the software packages to employ them appropriately and effectively. Further, the fragmented nature of the process results in maintenance problems and increased processing time.
THE IDEAL HTS DATA ANALYSIS METHOD
The ideal data analysis method would make use of all the screening results and thus base its conclusions about the potential of each family or individual compound on all the information present in the dataset. It would also take into account the presence of false positives and negatives and suggest potential false results for re-screening. Additionally, the ideal process would be fully automated, integrating all the subtasks required for a thorough analysis of an HTS dataset in a single package and thus eliminating the need for various packages and avoiding the problems of different data formats and software package compatibility.
More specifically, the envisioned HTS data analysis process should be able to select a subset of active compounds based on properties of the HTS dataset itself, including the presence of noise, and not on a user defined hard threshold. Appropriate algorithms that are able to detect the chemical families present and identify useful Structure-Activity Relationship (SAR) information should then process this subset of compounds. In order to identify all the significant SAR information the set of inactive compounds will have to be used as well. Once SAR information is available it should be used in conjunction with related corporate knowledge to evaluate the promise of all families and individual active compounds. Finally, the decision about the potential of individual compounds and families should be made and further exploration should commence.
PROPOSED METHODOLOGY GUIDELINES
Bioreason has adopted proven tools and methods from the computational intelligence field. Our methodology is based on automated decision support system concepts summarized by three main components:
Capture and automate the expertise of human analysts
Make use of recent advancements in the Knowledge Discovery and Data Mining fields and other branches of computational intelligence
Integrate expert and data driven processes into one system
CAPTURING HUMAN EXPERTISE
In order to capture expertise on screening data analysis, human experts assigned with the task of identifying leads in screening data were interviewed and consulted repeatedly. These experts were also involved in the process of defining the specifications during the design and implementation of the proposed decision support system. The majority of them described the process used as following:
Identify a subset of the hit list of manageable size, commonly the x most active hits or by use of a data reduction method such as clustering
Organize data in structural families of compounds
Look for Structure-Activity Relationships
Combine information to make hypotheses about which compounds to screen next, what would make compounds more or less active and what mechanisms of action the active compounds share
BUILDING AN AUTOMATED DECISION SUPPORT SYSTEM
Our automated decision support system was designed based on reasoning processes identified from experts in screening data analysis. A number of modifications were made to accommodate the deficiencies of computerized systems and capitalize on their strengths, especially their ability to analyze large amounts of data. Our modified process is defined in more detail below.
IDENTIFYING THE ACTIVE SUBSET OF THE HIT LIST
Instead of resorting to historical reasons for defining the activity threshold or arbitrarily picking a pre-defined number of the most active compounds imposed by the throughput of the process used, one can employ simple statistics to achieve better understanding of the data. This increased understanding enables a biological activity threshold selection that is less sensitive to biases. For example, in Figure 1, a bimodal distribution could be hypothesized for the readout and a good choice for the activity threshold could be values 14 or 15. By coupling data understanding with the subsequent usage of a fully automated process and scalable algorithms, lower thresholds of biological activity can also be selected. With a more robust analysis process, lower activities can be included in the initial analysis of the biological data as well. With this in mind, a researcher could choose to use a threshold of 10 or 11 for the screening data of Figure 1.
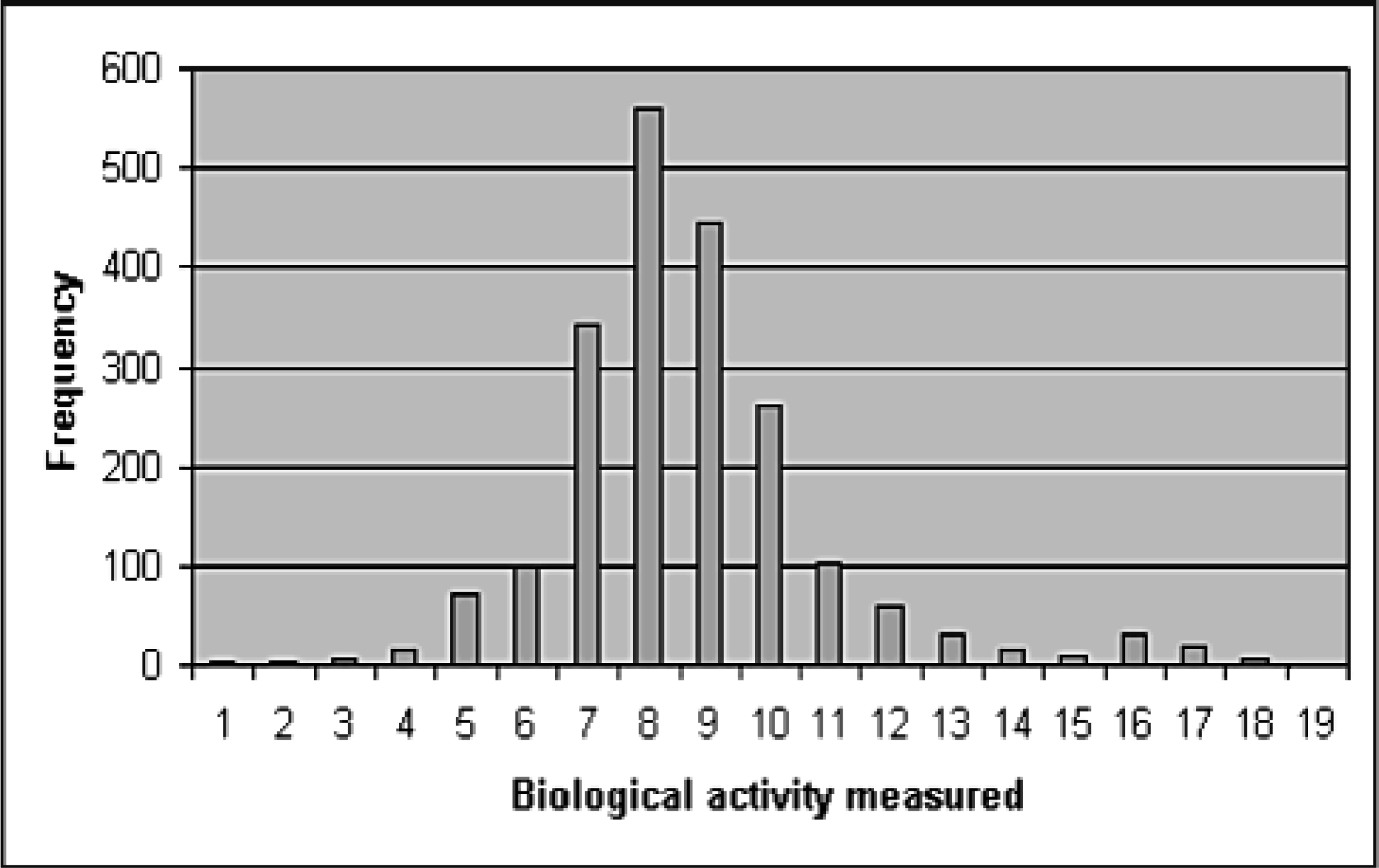
Typical screening dataset activity distribution.
ORGANIZING COMPOUNDS IN STRUCTURAL FAMILIES
Organizing compounds in groups, or clusters, based on representations of their chemical structure or biological characteristics is a subject well researched. 2,4,5 However, organizing compounds in chemical families based on common structural characteristics, e.g., chemical substructures or scaffolds, goes a step beyond.
Phylogenetic-Like Trees are data structures designed to represent the chemical family characteristics of the compounds of a dataset in a hierarchical manner. These trees are automatically constructed from a given screening dataset to organize molecules into structurally-similar groups/families, and to characterize those families which have one or more substructures shared by all the compounds in the structural family. Each tree is constructed based on the active compounds in the dataset and thus produces a set of structural families that are biologically interesting for the specific assay. The branches of the tree are extended level by level as the automated analysis proceeds, thus capturing finer and finer levels of detail about the family characteristics. After construction, the nodes of the tree are populated with inactive compounds that share the node's substructure. The nodes are also characterized with the usage of the properties of the compounds they contain via the application of statistical and expert driven methods.
The main goal for organizing data in this manner is to capture all significant chemical information from an HTS dataset in natural chemical family lineage. This organization helps focus the attention of the user on the interesting and relevant parts of chemistry space for that particular assay. The association of highly similar inactive compounds in each node and the ability to compute or import properties and characteristics of the compounds in each node make this data structure an excellent launching point for subsequent post-processing and knowledge extraction. Figure 2 shows a fragment of a fictional Phylogenetic-Like Tree.
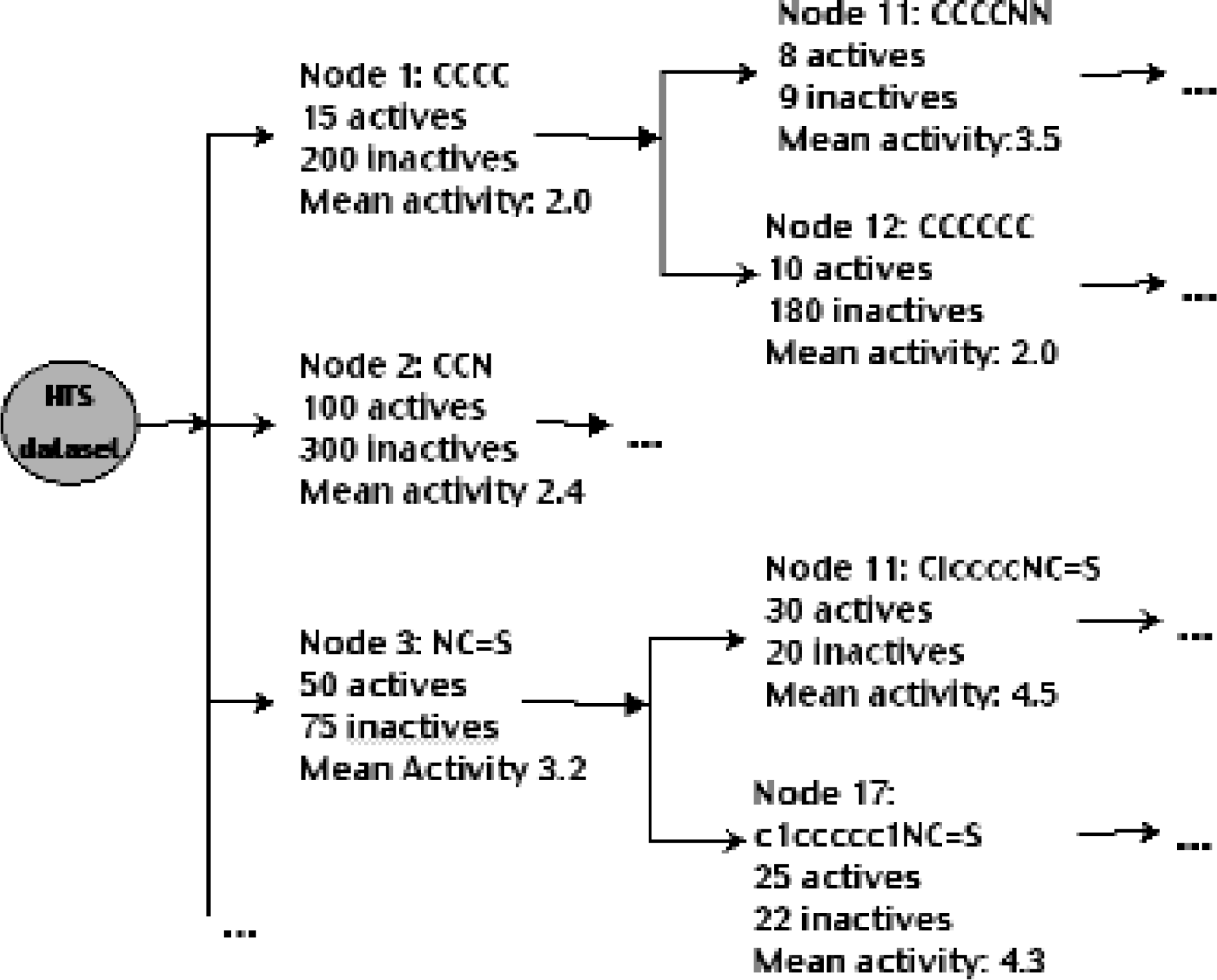
A fragment of a fictional Phylogenetic-Like Tree. Nodes of the tree fragment are shown to contain the substructure defining the node (in Smiles language notation), the number of active and inactive compounds sharing that substructure and the mean activity of all the compounds in the node. A typical Phylogenetic-Like Tree node contains additional properties obtained via the application of statistical and expert driven methods on the characteristics of the compounds it contains.
LOOKING FOR STRUCTURE-ACTIVITY RELATIONSHIPS
SAR identification is facilitated by the existence of a Phylogenetic-Like Tree. In this context it is reduced to a thorough exploration of the tree for potency enhancing and decreasing substructures. The hierarchical nature of the tree allows direct comparisons between parent nodes and their children for substructure changes that resulted in a significant increase or decrease of activity. Similarly, the nature of the tree allows easy comparisons among sibling nodes for better understanding of the effect that different substructure substitutions have on the parent family scaffold.
EXTRACTING KNOWLEDGE
The construction and exploration of a Phylogenetic-Like Tree results in a significant amount of information about the chemical families of interest and SAR present. All this information is used to prioritize the chemical families for follow-up studies. The prioritization is performed by a knowledge-based system containing rules defined in close cooperation with human experts. Classes with a higher potential for increasing potency, with SAR, may be given higher priority. Classes with minimal SAR may be given lower priority. Indications for potential false positives and false negatives are also available.
Bioreason's DrugPharmer™ is an interactive decision support interface employing an intuitive format to enable the user to:
assess and evaluate extracted knowledge
inspect data to verify the validity of extracted knowledge
query the raw data or the mined information
rapidly detect and focus on interesting and useful information
CONCLUSION
Informed and rapid decision making is highly desirable in the modern drug discovery process. In the case of analyzing HTS and uHTS screening data the current, human expert driven process is facing serious scalability problems. As a result the bulk of information (i.e. compounds with low or no biological activity measured) generated by the screening process is omitted and the time required for analysis has increased.
A way of alleviating this problem is to employ automated data analysis methods in the form of decision support systems. Such systems can emulate the data analysis process of human experts, apply that process without biases on the whole screening dataset in short periods of time and scale up to large amounts of data with relative ease. Human experts, while still having full control of the data analysis process, are then able to invest more time in exploring the knowledge extracted in the form of lead families and individuals, related SAR info, data validity info, etc., and thus make important decisions quicker and with more confidence.
ACKNOWLEDGEMENTS
The author would like to thank Drs. Susan I. Bassett and Bobi K. Den Hartog of Bioreason, Inc. for their useful suggestions and comments.