
Abstract
Abstract
Purpose
Synovial sarcoma (SS) is an aggressive soft-tissue tumor noted for late local recurrence and metastasis. This study investigates the long-term outcome of SS in patients of pediatric age and evaluates potential prognostic factors for SS.
Methods
We performed a retrospective review of 13 SS cases in patients younger than 20 years at the time of diagnosis who had a minimum follow-up of 10 years. The mean follow-up for living patients (n = 8) was 20.1 years (12.1–27.6) and for nonsurvivors (n = 5) 4.9 years (range: 2.6–9.3). Nine patients had unplanned excisions (69%), of which 6 (67%) were performed prior to their referral. Re-excisions were necessary in all 13 patients. The factors sex, tumor site, tumor size, tumor grade, histological subtype, fusion type, and type of treatment were evaluated for their prognostic value.
Results
Only 2 patients (15%) met the criteria of adequate tumor treatment. Overall, the 5- and 10-year survival rates were 77 and 61%, respectively. The mean time until a local recurrence (n = 5) was 3.2 years (range: 0.7–10.2), while there was a mean time of 2.1 years until the occurrence of late metastases (n = 5; range: 0.8–4.8). A high tumor grade and having a tumor in the trunk were adverse factors in terms of overall, local recurrence-free, and metastasis-free survival. Patients with wide resections or amputations had fewer local recurrences than patients with marginal or intralesional resections.
Conclusion
Inadequate primary excision of SS results in incomplete excision in the majority of cases. The tumor site, size, and histological grade should be considered when determining a risk-adapted treatment for SS, and wide surgical excision is the surgical intervention of choice. While local recurrence and late metastases appear to occur after a shorter time period in pediatric patients than in adults, in view of the tendency for late recurrence and metastasis with SS, follow-up should be at least 10 years.
Introduction
Synovial sarcoma (SS) is a malignant soft-tissue sarcoma that accounts for approximately 5–10% of all soft tissue sarcomas. Although the incidence rate of SS peaks in the third decade of life [1], 30% of SS cases occur in patients younger than 20 years of age [2].
Delay in diagnosis and treatment is common, because SS remain unrecognized for a long time. A significant number of excised lumps after an unplanned excision unexpectedly turn out to be sarcomas [3]. Most present with a painful lump which gradually grows over time. In pediatric cohorts, survival rates of up to 75–84% [2, 4–9] have been reported. These are superior to those reported in adults (survival rates ranging from 25–71%) [4, 10–14].
Based on morphology, SS is divided into two types histologically: biphasic and monophasic. Histogenetically, SS is characterized by a specific chromosomal translocation, t(X;18) (p11;q11), and the resultant fusion gene. There are two major subtypes of the fusion gene: synovial sarcoma translocation SSX1 and SSX2 [15, 16]. Prognostic factors in SS remain a controversial topic [13]. Tumor stage [2], male sex, trunk-related tumors [5, 8], large tumor size [2, 4, 5, 7, 9, 17, 18], and high tumor grade [7, 14] have all been associated with an adverse outcome of SS.
Surgery is the basic treatment for SS, and obtaining adequate margins is its principle aim [10, 18]. The current standard treatment for SS is wide resection followed by polychemotherapy with or without irradiation [11, 19–21]. While radiotherapy has a role to play in improving local control of the tumor and, subsequently, overall survival [22], the role of chemotherapy in the treatment of SS remains controversial [7, 23]. Due to the rarity of SS, there have been no randomized trials studying the use of chemotherapy in the treatment of SS. However, as with other soft tissue sarcomas, surgical resection (with or without adjuvant radiotherapy) is often accompanied by chemotherapy [6, 24] The use of adjuvant therapy has even been suggested in children that undergo nonradical surgery [2, 6], although the value of this approach in terms of overall survival still remains to be studied.
The tendency for local recurrence (LR) and late metastases with SS is well known and reflected by the marked differences between short- and long-term survival [17, 25]. Most published reports include only single patients with long follow-up times. The data on long-term outcomes is limited to one report published by Guadagnolo et al., which had a median follow-up of 13.2 years (range: 2.4–37.5 years). Nineteen percent of the patients in that study were of pediatric age (n = 29) [26]. Based on our clinical observations indicating the prevalence of late occurrences of metastases and local recurrence in SS patients, we hypothesized that a long-term follow-up study was essential to properly design post-surgical care for these patients. Therefore, we designated a minimum follow-up of 10 years as the main inclusion criterion for participation in this study.
Patients and methods
After gaining the approval of the local ethical committee, we performed a retrospective, multicentric review of all SS cases recorded by the Swiss University tumor centers. The study was conducted under the guidance of the Department of Paediatric Orthopaedics at the Children's University Hospital in Basel with the participation of the Institute of Pathology of the University Hospital Basel, Lausanne, and Zurich Enge, the University Hospital Balgrist Zurich, and the Paediatric Orthopaedic Department at the University Hospital in Geneva. We included patients ≤20 years of age at the time of the diagnosis of SS and a follow-up of at least 10 years. Thirteen (6 male, 7 female) of the 62 patients that had been treated for SS between 1968 and 1999 met these criteria and were included in the study. Patient and tumor data were retrieved from the records of the participating hospitals and pathological institutes.
Eight of the 13 (61.5%) were surgically treated at outside institutions (i.e., not one of the aforementioned university centers) prior to their referral. All but 2 patients (84%) had symptoms suspicious for malignancy (i.e., pain and/or increase in size). In addition to pain and swelling, 2 patients with a tumor in the popliteal fossa and 1 with a tumor at the elbow showed a decreased range of motion at the affected joint as a primary symptom.
Only 4 patients had a planned biopsy. There were 9 patients with unplanned excisions (69%), of which 6 (67%) were performed prior to the referral to one of the participating centers. Re-excisions were necessary in all 13 patients. There were 2 amputations, one after an incisional biopsy of a tumor in the popliteal fossa and the other after multiple resections of a SS at the elbow (see Table 1). Only 2 patients met the criteria of adequate tumor treatment with a planned biopsy, followed by a wide resection or a resection with clear margins.
Patient characteristics and tumor data
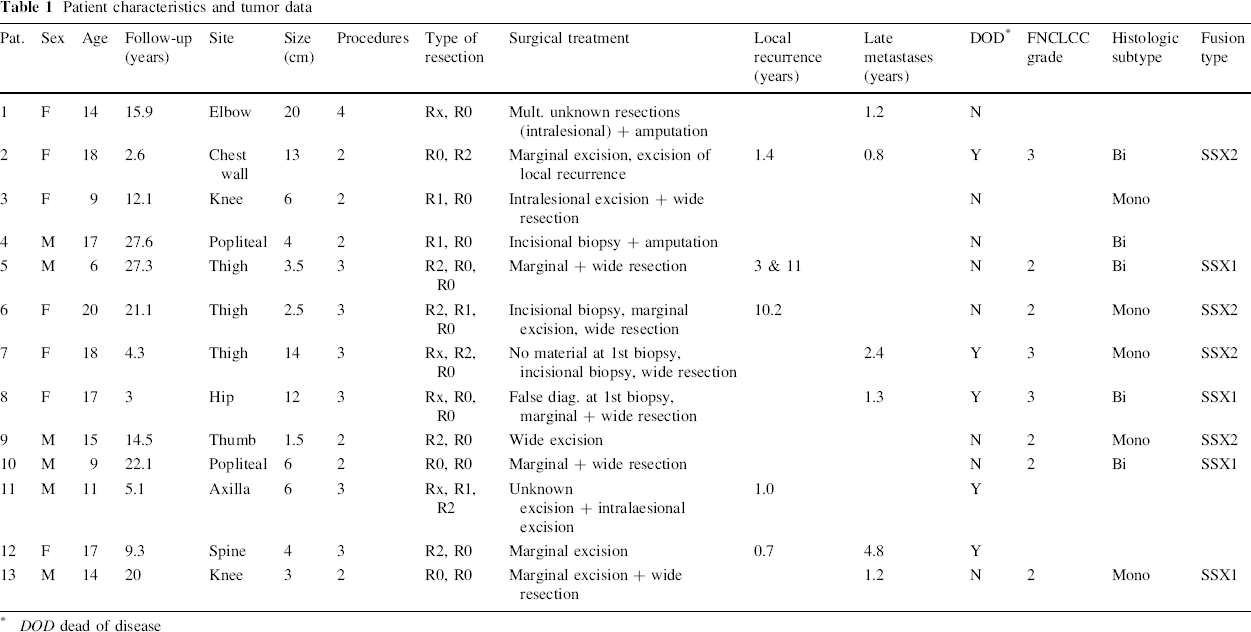
DOD dead of disease
Three patients had a postoperative chemotherapy. Nine patients were treated with neoadjuvant chemotherapy, of which 7 received a combination of Adriamycin and Ifosfamid. Four patients received an additional agent (3 vincristine, 1 bleomycin). One patient underwent preoperative radiotherapy of 45 Gy to downsize a large 14 cm tumor with infiltration of blood vessels to make it more accessible to the wide en-bloc resection ultimately performed. The average radiation dose in patients with adjuvant radiotherapy (n = 9) was 54 Gy.
The mean follow-up time was 14.2 years (range: 2.6–27.6). The mean follow-up was 20.1 years (range: 12.1–27.6) for living patients (n = 8) and 4.9 years (range: 2.6–9.3) for nonsurvivors (n = 5). After written informed consent was obtained, all living patients without adequate clinical follow-up were invited to undergo clinical examination, which included an MRI of the site of the primary tumor, a chest X-ray, and an ultrasound of the abdomen and regional lymph nodes [27].
Two reference pathologists (LG, GJ) reviewed the histological slides and blocks and verified the histological diagnosis. Tumor grading was based on the French Federation of Cancer Centers Sarcoma Group (FNCLCC) grading system, a three-grade classification that determines a score based on tumor differentiation, mitotic index, and tumoral necrosis [27]. Paraffin-embedded specimens of 8 patients were analyzed for the type of SYT-SSX fusion by reverse-transcriptase polymerase chain reaction, as previously described [14, 28]. The factors tumor site (limb-based and trunk-related), sex, tumor size (≤5 cm vs. >5 cm), FNCLCC grade (G2 vs. G3), histological subtype (bi- vs. monophasic), fusion type (SSX1 vs. SSX2), type of surgical and adjuvant treatment modality, and tumor margins were evaluated for their potential prognostic value.
The statistical software package SPSS (Statistical Product and Services Solutions, version 17.0, SPSS Inc., Chicago, IL, USA) was used for all data analysis. Overall survival (OS), local recurrence-free survival (LRFS), and metastasis-free survival (MFS) at 5 and 10 years were obtained according to the Kaplan–Meier estimate [29]. Comparisons were tested for statistical significance using the log-rank test [30]. The results of significance tests were expressed in P values, with a P value of <0.05 indicating statistical significance.
Results
Patient and tumor data are summarized in Tables 1 and 2. The mean age at diagnosis was 14.2 years (range: 6–20).
Patient characteristics (age, sex, tumor site, tumor size, tumor grade, subtype, SSX1 + 2, type of treatment)
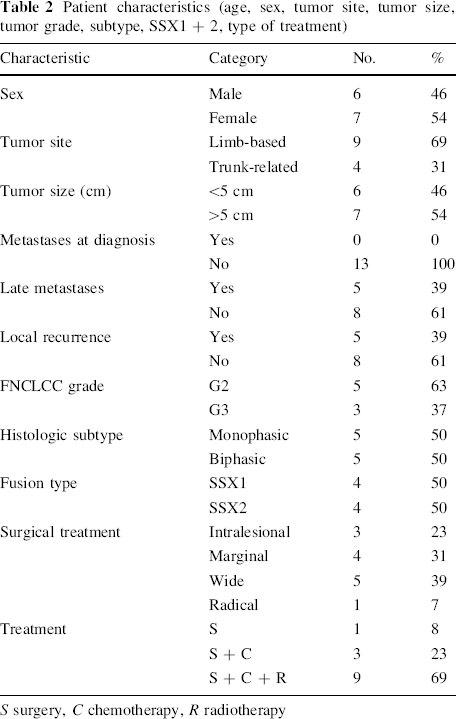
S surgery, C chemotherapy, R radiotherapy
One patient had surgery without additional treatment, 3 had chemotherapy after surgery, and 9 patients underwent surgery following neoadjuvant chemotherapy and radiotherapy. There was no significant difference in overall survival between the latter two groups (P = 0.136).
At the time of the last follow-up, 8 patients (61.5%) were still alive and 5 (38.5%) had died due to a tumor-related cause. As 2 patients died from the disease ≥5 years post-surgery, the 5- and 10-year overall survival rates were 76.9 and 61.5%, respectively (Fig. 1). There was no significant difference in overall (P = 0.13) and local recurrence-free (P = 0.662) survival in terms of sex. The variables histological subtype (monophasic vs. biphasic) and fusion type (SSX1 vs. SSX2) did not significantly influence the overall survival (P = 0.426 and 0.508, respectively).
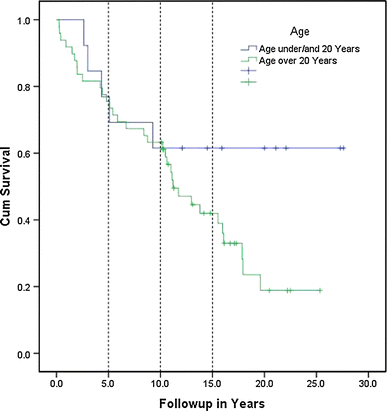
Overall survival: 5- and 10-year overall survival rates
Of the 9 patients (69.2%) with limb-based tumors, 2 patients had tumors in the upper extremities and 7 patients had them in the lower extremities. Patients with limb-based tumors (n = 9) had significantly better overall (P = 0.001) and metastasis-free (P = 0.07) survival than patients with trunk-related SS (n = 4). The mean tumor size was found to be 7.3 cm (range: 1.5–20 cm). There was no difference in the overall survival between patients with tumors ≤5 cm (n = 6) and those with tumors >5 cm (n = 7; P = 0.107). Patients with G2 tumors (n = 5) had significantly better overall and metastasis-free survival than those with G3 tumors (n = 3; P = 0.004; Fig. 2).
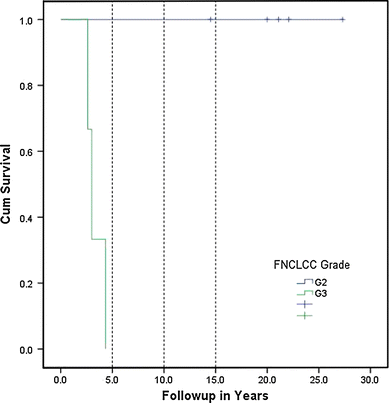
Tumor grade
Patients with an intralesional resection (n = 3) had significantly poorer overall survival than patients that had a wide resection, an amputation, or a marginal resection (P = 0.021). There was no difference in terms of overall survival in patients that had wide resections with clear margins compared to those with a marginal or intralesional resection (P = 0.9). However, only patients with intralesional (n = 2) and marginal resections (n = 3) were found to have local recurrences (n = 5, 39%).
None of the patients had metastases at the time of diagnosis. Five patients developed late metastases after surgical intervention, and the mean time to develop metastases was 2.1 years (range: 0.8–4.8 years). At the time of the last follow-up, 8 patients were alive without evidence of disease. The MFS at 5 years was 61.5% and remained constant at 10 and 15 years (Fig. 3). Patients with late metastases (n = 5) had a significantly worse overall survival at 5 and 10 years than those without (n = 8; 40 vs. 100% at 5 years and 20 vs. 87.5% at 10 years; P = 0.01). Further, patients without local recurrence (n = 8) had a significantly better outcome in terms of MFS (P = 0.01).
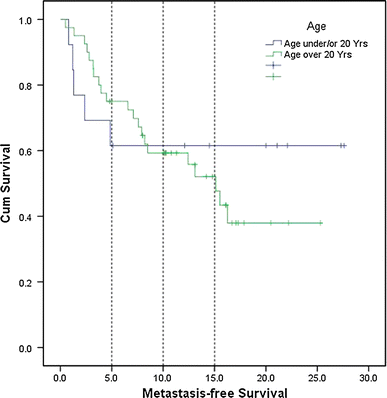
Metastasis-free survival
The mean time until a LR occurred was found to be 3.2 years (range: 0.7–10.2). There was no significant difference in terms of overall survival in patients with a local recurrence compared to those without (75 vs. 40%; P = 0.27). There was no significant difference in terms of LRFS in patients ≤20 years compared to those >20 years (P = 0.41; Fig. 4). No local recurrences occurred in patients with wide resections or amputations. The type of resection (P = 0.016) and the tumor site (limb-based vs. trunk-related; P < 0.01) had a significant influence on the occurrence of a local recurrence.
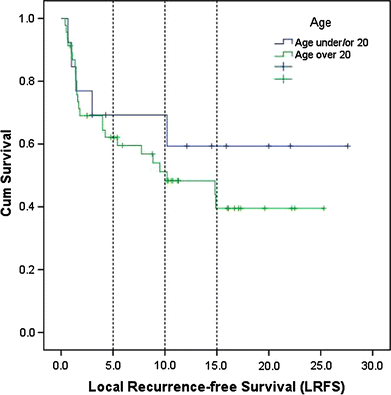
Local recurrence-free survival
Discussion
In the literature, the roles that several factors have on the outcome of SS in children remain controversial, especially with respect to the long-term outcome. Thus, we designed a study that had a minimum follow-up of 10 years to try to elucidate the importance of these factors. The present study has several limitations, mainly sample size. As in other studies of SS, the rarity of the disease led to a small study population (13 patients in this case). However, considering that this was a multicentric study with the participation of all University Tumor Centers of our country, this number reflects the total number of pediatric and adolescent cases of a whole country. The defined minimum follow-up time of 10 years is another strong point of the study.
Neither the histological subtype nor the type of translocation (SSX1 vs. SSX2) had any significant effect on the outcome of SS [14]. This is in accordance with our study of adult patients [27]. As SS is characterized by the specific chromosomal translocation t(X;18) (p11;q11), immunohistochemical and cytogenetic studies are excellent tools for diagnosing SS. However, they do not help with the process of deciding on the appropriate surgical and/or adjuvant treatment [14, 27, 28]. In SS, the knowledge of which fusion gene (SSX1 or SSX2) the tumor possesses may be of vital importance, as the fusion protein (the product of the fusion gene) is a promising target for therapeutic innovation [27].
In a previous study by Guillou et al., the histological grade is described as a significant factor associated with a poorer outcome in regards to overall, metastasis-free, and local recurrence-free survival in high-grade tumors [14]. Similar findings were also found in a study of 37 children and adolescents [7]. Our data indicating that patients with G2 tumors had significantly better overall and metastasis-free survivals than those with G3 tumors (P = 0.004) is in accordance with these findings (Fig. 2). Therefore, the initial biopsy is not only the keystone in defining the diagnosis; it is also of major importance in clinical decision-making, as a high histological grade should skew the decision towards a more aggressive approach.
As in other soft-tissue sarcomas, the quality of the surgical operation is crucial. Obtaining adequate surgical margins is essential to gain tumor control, and is strongly influenced by the type of healthy tissue surrounding the tumor [10, 18]. This is supported by our results, which showed that patients with a primary wide resection had a better outcome in terms of OS, MFS, and LRF, while patients with initial marginal or intralesional resections had a significantly worse prognosis than the other patients.
In our study, patients with limb-based tumors had significantly better overall (P = 0.001) and metastasis-free (P = 0.07) survivals than patients with trunk-related SS (n = 4). This corresponds to results published in the literature [6, 7, 24]. A possible explanation for this is that tumors located in the trunk tend to become clinically apparent at a later stage of tumor growth, leading to a delay in diagnosis. Additionally, with tumors in the trunk, it is more difficult to achieve an R0 surgical resection without damaging viable neurovascular structures, and distant pulmonary metastasis appears to occur more often [2]. Therefore, the tumor site should be considered when deciding on a risk-adapted treatment strategy for SS.
In contrast to several published studies [2, 4, 5, 8, 14, 17, 31, 32], our study did not demonstrate that tumor size has an influence on the outcome of SS (P = 0.107). In our previous study, and in the literature, size was found to have a significant effect on metastasis-free, overall, and local recurrence-free survivals [2, 4, 27].
The high rate of adjuvant chemotherapy in our patients (n = 12, 92%) compared to the adult patients in our previous study (46%) gives rise to the question: does chemotherapy have a prognostic influence on SS? On the other hand, it also leads one to question whether young patients have been overtreated. However, the diversity of treatment regimens used in our patients, and the fact that almost all patients received chemotherapy, makes it impossible to clarify the influence of chemotherapy on the clinical outcome here.
Although the difference in overall survival between patients with local recurrences and those without (75 vs. 40%) did not reach statistical significance (P = 0.27), we still think there is a clinical relevance to this finding. The small size of the study population might be responsible for the lack of significance. Another possible reason for the lack of a difference may be the fact that the time needed to treat the tumor seems to be shorter in children than it is in adults. Consequently, tumors tend to be treated at a smaller size.
As opposed to our previous study with adult patients [27], there were no young patients with metastases at the time of diagnosis. Late metastases tended to occur earlier in children than in adults. However, pediatric patients had better long-term overall and metastasis-free survivals than adult patients (Fig. 3). This is in accordance with Sultan et al., who demonstrated higher cancer-specific 5- and 10-year survivals for children/adolescents (83 and 75%) than in adults (62 and 52%) [27, 32]. In accordance with their findings, we also observed that children present with better clinical features, such as smaller primary tumors and more limb-based tumors.
We acknowledge that many of the patients in our study received what is now considered to be inappropriate and substandard therapy. This is something that our study revealed during the course of the data collection. We confirm the observation that the unplanned excision of SS results in incomplete excision in the majority of cases [3]. However, we are convinced that this reflects the clinical reality for the majority of patients presenting to the doctor with “a lump that has grown over time.” Only 4 of the 13 patients (31%) had a planned biopsy. There were only two patients that met the criteria of adequate tumor treatment with a planned biopsy, followed by a wide resection or a resection with clear margins. This depicts the reality that the standard therapy in the presented study population is a substandard therapy—a key result of our study.
To prevent the incomplete excision of a sarcoma, a carefully planned biopsy should always be the initial surgical procedure in patients with a suspicious soft-tissue mass. Wide surgical excision is the treatment of choice for SS. The tumor site, size, and histological grade should be considered when determining a risk-adapted treatment for SS. In view of this tumor's tendency for late recurrence and metastasis, follow-up should be at least 10 years.
Footnotes
Acknowledgments
The authors wish to acknowledge Ms. Petra Huber for her assistance in the acquisition of the pathologic material, and the Susy Rückert Foundation for funding the study. We also thank Bioscience Writers, Houston, Texas, for the editing of the manuscript.
None.