
Abstract
Purpose
Greig cephalopolysyndactyly (GCPS) (OMIM 175700), a rare autosomal dominant disorder, is characterized by a distinct combination of craniofacial, hand and foot malformations. The hand and foot malformations often require orthopedic assessment and treatment. The disorder is caused by point mutations or deletions in the GLI3 gene, located on chromosome 7p14.3. Herewith, we review the hand and foot malformations in a cohort of 13 patients referred for genetic testing.
Methods
We reviewed the medical files of 13 patients with GCPS seen at the Center for Human Genetics in Leuven between 2003 and 2005. Clinical, molecular and radiological findings, when available, were recorded.
Results
We identified six different point mutations in the GLI3 gene, two microdeletions and three larger chromosomal deletions. In the hands, preaxial polydactyly was never observed, but the malformations included postaxial polydactyly, broad thumbs, clinodactyly of the thumbs and various degrees of syndactyly. In the feet the spectrum of malformations included preaxial polydactyly, postaxial polydactyly, different degrees of syndactyly and broad halluces. Syndactyly of the toes and hallux abnormalities were present in all patients. Most frequently, syndactyly was present between toes 1–2–3. The broadening of the hallux was either due to a complete or partial duplication of the first toe or to broadening of the distal phalanx. Mental retardation was found in three cases and was associated with a large chromosomal deletion of the GLI3 region.
Conclusion
We found the classic hand and foot malformations associated with GCPS in our cohort of patients. Patients with a large chromosomal deletion had mental retardation, but no structural brain anomalies were found.
Introduction
Antero-posterior patterning of the limbs is controlled by the zone of polarizing activity (ZPA), a region of posterior limb bud mesoderm. In the ZPA, the key mediator is Sonic Hedghog (Shh). Genetic studies in mice and chickens have demonstrated that Shh and Gli3, one of its downstream targets, are important in the development of the digits, more specifically in the regulation of digit number and digit identity. Ectopic implantation of the ZPA or Shh in the anterior part of the limb bud of chick embryos induces mirror-image duplications [1]. Gli3, the homologue of the Drosophila cubitus interruptus gene, encodes a transcription factor that plays a crucial role in antero-posterior patterning of the limb bud. The extra toes (Xt) mouse mutation creates a Gli3 null allele, and Xt heterozygotes are characterized by preaxial digit alterations. Homozygote Gli3 −/− mice have severe polydactyly [2–4]. Moreover, mice lacking Gli3 and Shh (Shh −/− Gli3 −/− ) have 6–11 syndactylous digits per limb [5]. Gli3 and Shh thus both act in normal patterning of the limb by inhibiting the development of more than five digits.
The importance of Gli3 and Shh in digit formation is also reflected in human polydactyly syndromes. Mutations in a SHH enhancer have been implicated in isolated preaxial polydactyly [6]. Point mutations or deletions in the GLI3 gene cause Greig cephalopolysyndactyly (GCPS; OMIM 175700) [7–10]. GCPS was first described in 1926 by Greig [11]. It is a rare autosomal dominant disorder characterized by abnormal craniofacial and limb development. Craniofacial abnormalities include frontal bossing, hypertelorism, macrocephaly and a broad nasal bridge. In the hands postaxial polydactyly and (less frequently) preaxial polydactyly can be observed. The postaxial polydactyly usually manifests itself as a pedunculated postminimus. Thumbs are usually broad, and the terminal phalanx can be bifid. In the feet there is often duplication of the hallux (preaxial polydactyly) combined with syndactyly of toes 1, 2 and 3. Very seldom, postaxial polydactyly is seen in the feet [12]. After the original description by Greig in 1926, several other authors have contributed to the delineation of the characteristic features of this syndrome [13–31].
In this study we review the clinical, radiological and molecular features of 13 individuals with GCPS with established GLI3 mutations. We specifically focus on the orthopedic abnormalities in the hands and feet.
Materials and methods
We reviewed the medical files of 13 patients with established GLI3 mutations seen at the Center for Human Genetics in Leuven between 2003 and 2005. Clinical, molecular and radiological findings, when available, were recorded. Molecular analyses included standard karyotyping and GLI3 mutation analysis. Amplification of the 14 translated GLI3 exons and their adjacent intron sequences was performed using genomic DNA isolated from blood as template with previously described primer pairs [7]. For single-strand conformation analysis (SSCA), PCR products were diluted in glycerol/formamide containing buffer and separated following denaturation on native 12% acrylamide gels at 10 or 20°C (600 V) for 1.5 h. Conformers were visualized by silver staining. SSCA banding patterns deviating from the wild-type pattern were compared with those from the control population (n = 100). PCR fragments generating deviant SSCA bands not representing polymorphisms were sequenced following standard procedures to identify the underlying change in DNA composition. To detect microdeletions or duplications within the GLI3 gene region that escape detection in the SSCA as well as DNA sequencing, the copy number of individual exons was determined by multiplex amplifiable probe hybridization (MAPH) [32]. The probes used for quantitative recovery after their hybridization to immobilized patient DNA were exonic PCR products cloned into the pCR2.1TOPO vector (Invitrogen Inc., Merelbeke, Belgium). To generate these PCR products, translated GLI3 exons were amplified with the same primers and conditions as for SSCA. One unique probe each from chromosomes 5 and X was taken as control.
This study was approved by the Ethical Board of the University Hospital in Leuven, Belgium.
Results
Familial inheritance was observed in five individuals belonging to two independent families. Family A consists of two affected individuals (PARU and BRER, his mother). Family B is a three-generation family with three affected individuals (SNNE, MEHA, DRGO). The remaining eight GCPS patients represent sporadic cases.
Table 1 summarizes the clinical data of the 13 GCPS patients. All patients had a combination of hand and/or foot malformations associated with one or more facial features typical for GCPS: a high, broad forehead, hypertelorism, a broad nasal bridge and frontal bossing (Fig. 1).
Schematic overview of the clinical features observed in the patients with GCPS
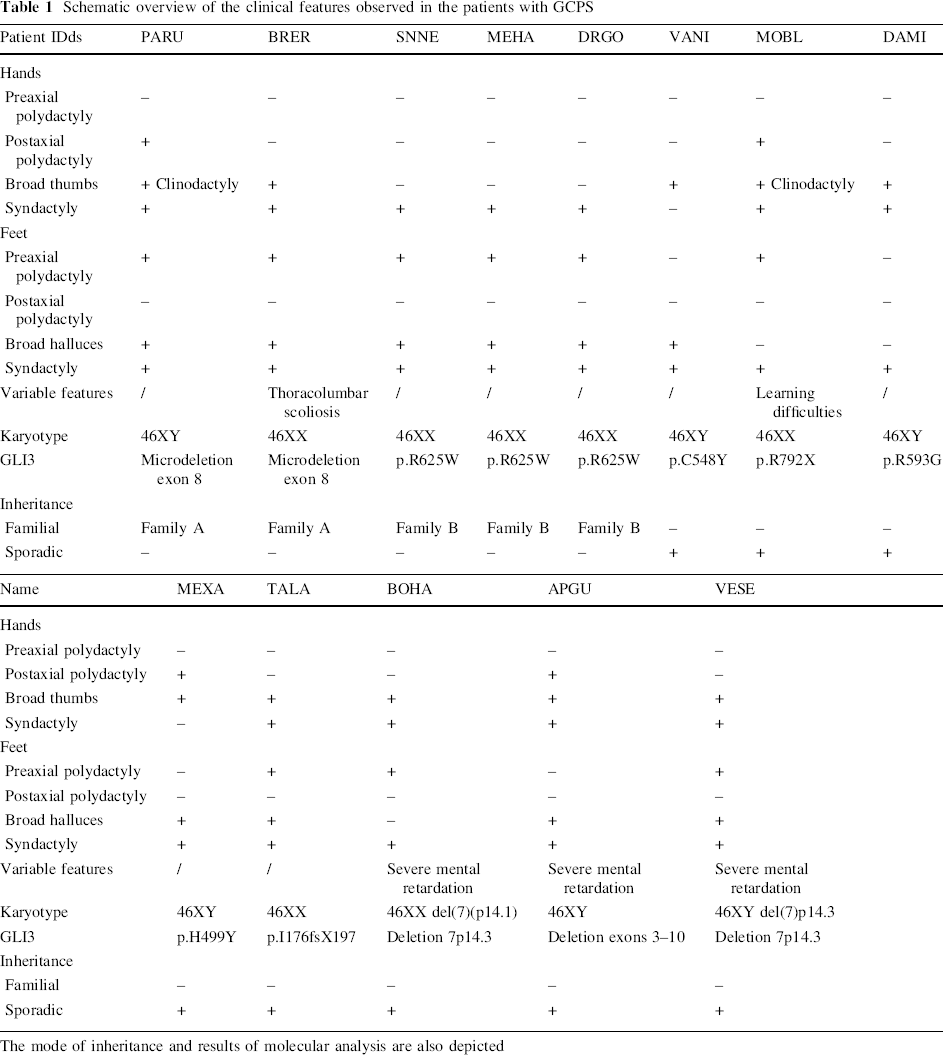
The mode of inheritance and results of molecular analysis are also depicted
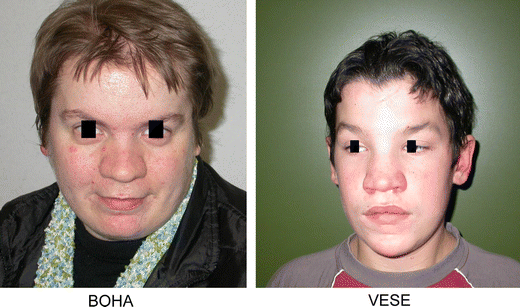
Typical craniofacial appearance of GCPS patients: frontal bossing, hypertelorism, a high forehead and a broad nasal bridge
Hand malformations (Fig. 2)
Syndactyly of the fingers was the most frequent finding in the hands (11 cases). The degree of syndactyly varied from discrete cutaneous syndactyly to syndactyly of the entire fingers with bony involvement. Postaxial polydactyly was observed in four cases and consisted of a pedunculated postminimus. Preaxial polydactyly of the hands was never seen, but a broad thumb was a frequent finding (nine cases). Radiographically, broadening of the thumb can be due to duplication of the distal phalanx, the presence of a delta phalanx or broadening of the distal phalanx. In one patient, broadening of the thumb was caused by an irregular epiphysis of the distal phalanx (MOBL). In two cases (PARU and VANI) clinodactyly of the thumb was observed. Radiographically, this feature corresponded to a malformation of the proximal phalanx.
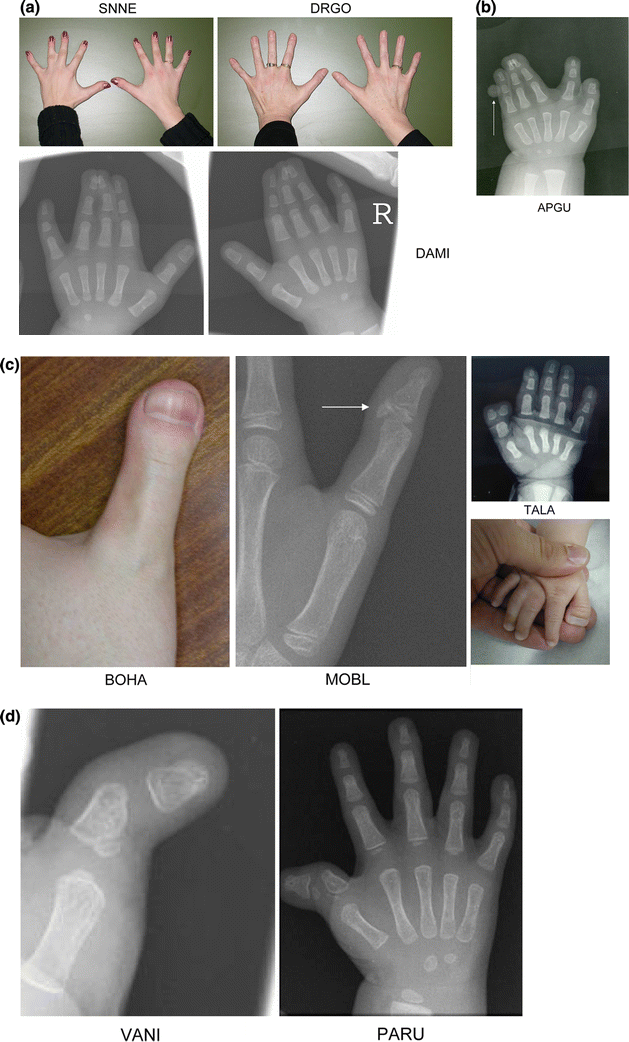
The spectrum of hand malformations in patients with GCPS.
Foot malformations (Fig. 3)
Syndactyly of the toes was a consistent finding in all examined cases. Most frequently syndactyly between toes 1–2–3 was observed (seven cases). Three cases had syndactyly between toes 2 and 3, two patients had syndactyly between toes 1–2 and one patient had syndactyly of toes 1–2–3–4. Postaxial polydactyly was never observed, but preaxial polydactyly was noted in nine patients. The polydactyly can be partial or complete. Broadening of the hallux was noted in all patients. Radiographically, this was reflected as a partial or entire duplication of the distal phalanx associated with malformations of the proximal phalanx and the first metatarsal. In some cases a duplication or simple broadening of the first metatarsal was present. In MOBL the aberrant widening of the first metatarsal induced a medial deviation of the entire forefoot.
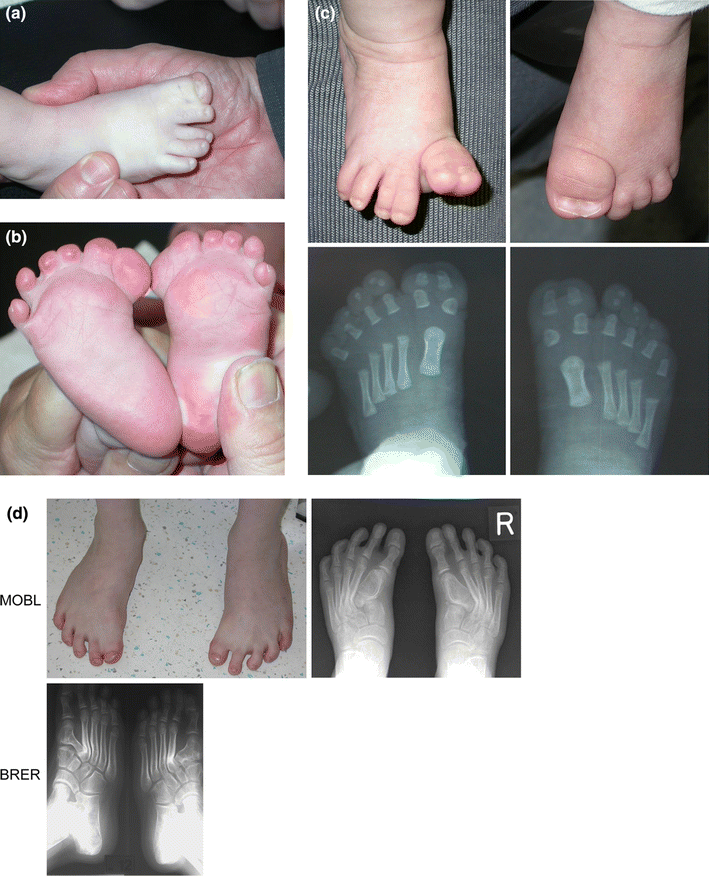
The spectrum of foot abnormalities in patients with GCPS.
Clinical features not typical for GCPS
A thoracolumbar scoliosis was observed in one patient (BRER). APGU, BOHA VESE had severe mental retardation and MOBL had learning difficulties.
Molecular analyses
Two patients had an abnormal karyotype: VESE and BOHA had a cytogenetically visible deletion of chromosome 7p, more specifically the 7p14.3 region that harbors the GLI3 gene. All other patients had a normal karyotype. SSCA and sequencing led to the identification of GLI3 point mutations in eight patients. We detected a total of six different point mutations. All mutations were present in a heterozygous state. The three affected members of family B carried the missense mutation p.R625W. Other missense mutations were also found in DAMI (p.R593G), VANI (p.C548Y) and MEXA (p.H499Y). A nonsense mutation p.R792X was present in MOBL, and a frameshift mutation (p.I176fsX197) was found in the GLI3 sequence of TALA. In the remaining three patients with a normal karyotype no GLI3 mutation could be identified. However, MAPH analysis in these patients revealed the presence of a microdeletion of exon 8 of GLI3 in BRER and her son PARU (family A) and a large deletion of exons 3 to 10 of GLI3 in APGU.
Discussion
In this report we present the clinical, radiological and molecular data of 13 patients with GCPS seen at the Centre for Human Genetics in Leuven, with specific attention for the malformations in the hands and feet, since these malformations are frequently a reason for corrective surgery. Polydactyly of the hands and feet is a cardinal feature of GCPS. In the hands postaxial polydactyly is frequently seen, whereas in the feet preaxial polydactyly is more common. Postaxial polydactyly in our patients always consisted of a small cutaneous tag on the ulnar side of the hand (pedunculated postminimus) and was in all cases removed shortly after birth. Preaxial polydactyly in the hands was never observed in our group of GCPS patients, but a broad thumb was present in nine of them. Clinical broadening or widening of the thumb correlated on radiographs with a broad distal phalanx, a bifid distal phalanx, the presence of a deltaphalanx or irregularities of the epiphysis. Abnormalities of the proximal phalanx were not frequent, but in two cases they resulted in clinodactyly of the thumb. At the feet preaxial polydactyly was present in nine patients. It was expressed as a broad hallux with broad and/or bifid nails or as an entire duplication of the hallux. Radiographically, broadening of the hallux was caused by partial or entire duplication of the distal phalanx. In some cases also the proximal phalanx was duplicated and in two cases we noted partial or entire duplication of the first metatarsal. In two cases the first metatarsal was grossly deformed creating an abnormal articulation with the base of the second metatarsal. Syndactyly of the toes and fingers was also a frequent finding. Mostly, the syndactyly was cutaneous, but occasionally bony fusions were present in the syndactylous web.
Mutations in the GLI3 gene and interruptions of the GLI3 gene by chromosomal deletions or translocations cause GCPS [7, 8, 10]. As mentioned above, the GLI3 protein is a zinc finger transcription factor that plays an important role in the SHH pathway. This pathway is not only important for antero-posterior patterning of the developing limb, but is also used to specify the neural tube, the craniofacial structures and many other structures. The important developmental function of GLI3 and its expression in different embryonic tissues is also reflected in the phenotypic features of other syndromes associated with GLI3 mutations. Apart from GCPS, GLI3 mutations can also result in Pallister-Hall syndrome (PHS, OMIM 146510) [33], characterized by central or postaxial polydactyly, hypothalamic hamartoma, pulmonary segmentation anomalies, bifid epiglottis or laryngeal clefts. GLI3 mutations have also been observed in different types of polydactyly, postaxial polydactyly type A1 (PAP-A; OMIM 174200), postaxial polydactyly type A/B (PAP-A/B) and preaxial polydactyly type IV (PPD-IV; OMIM 174700) [34–35], and the acrocallosal syndrome (ACLS, OMIM 200990) [36]. ACLS has several features in common with GCPS: polysyndactyly, macrocephaly and hypertelorism. Patients with ACLS, however, also have agenesis or dysgenesis of the corpus callosum, a feature not specific for GCPS. Mental retardation is always present in ACLS, and it can occasionally be seen in GCPS patients. Finally, ACLS is autosomal recessive [37], whereas GCPS is clearly inherited in an autosomal dominant fashion. We found a causative GLI3 mutation in eight cases. Two of these mutations have been previously described (p.R625W and p.R792X) [7, 16, 23], but the other mutations (p.C548Y, p.R593G, p.I176fsX197, p.H499Y) are novel. Five patients had a deletion of GLI3. In two cases the deletion was cytogenetically visible. One patient had a smaller deletion encompassing exons 3–10 of GLI3, and two patients had a microdeletion of exon 8. Interestingly, three patients had mental retardation. Patients with a small deletion of GLI3 (PARU and BRER) had a GCPS phenotype without evidence for developmental delay or mental retardation. GCPS patients with mental retardation were the ones who had large deletions of the region on chromosome 7p harboring the GLI3 gene. Mental retardation has been associated with GCPS [22, 24, 29, 31, 38], and as mentioned above, GCPS and ACLS have several overlapping features. None of our patients with large deletions had agenesis of the corpus callosum. Therefore they were not classified as ACLS, but as ‘severe’ GCPS cases. Johnston et al. [22] have designated this condition the ‘GCPS contiguous gene syndrome’: the large deletion on chromosome 7p removes, apart from the GLI3 gene, several other genes, some of which may be responsible for normal neurological development, hereby causing the observed mental retardation. Therefore, when the classic features of GCPS are associated with mental retardation, an MRI scan of the brain is recommended to exclude agenesis of the corpus callosum.
It should be noted that other syndromes can also be associated with pre- and postaxial polydactyly of hands and feet, and as such, they should always be considered in the differential diagnosis of GCPS. Central or postaxial polydactyly is typical for Pallister-Hall syndrome. Different types of polydactyly can be observed in postaxial polydactyly type A1 (PAP-A) (OMIM 174200), postaxial polydactyly type A/B (PAP-A/B) and preaxial polydactyly type IV (PPD-IV; OMIM 174700). Isolated preaxial polydactyly of the hands is caused by mutations in a regulatory element of the SHH gene [6]. All these syndromes, however, lack the typical craniofacial manifestations of GCPS. In the group of oral–facial–digital syndromes (OMIM 311200) a hand- and foot-phenotype similar to the one in GCPS can be seen associated with minor facial anomalies (frontal bossing, low-set ears, hypertelorism, broad nasal bridge, etc.). These patients also have distinct oral abnormalities (cleft or lobulated tongue, oral frenula, and/or cleft palate) not typical for GCPS. A facial appearance similar to GCPS can be seen in cranio-fronto-nasal syndrome (OMIM 304110). In this syndrome, females have frontonasal dysplasia, craniofacial asymmetry, craniosynostosis, bifid nasal tip, grooved nails, wiry hair and abnormalities of the thoracic skeleton, whereas males typically show only hypertelorism. Unlike GCPS, hands and feet are usually normal in cranio-fronto-nasal syndrome.
In conclusion we can say that GCPS is a rare autosomal dominant disorder. Nevertheless orthopedic surgeons should be aware of this syndrome since these patients are often referred for corrective surgery on the hands and/or feet. Since orthopedic surgeons are sometimes the first to see such patients, they should be know that the hand and foot malformations can occasionally be associated with brain abnormalities. Further investigations and genetic counseling are therefore mandatory both in sporadic and familial cases, especially since inter- and intrafamilial variability of the phenotype has been described [13, 14, 16, 18, 20, 25].
Footnotes
Acknowledgments
P. Debeer is a Senior Clinical Investigator of the Fund for Scientific Research, Flanders (Belgium).