
Abstract
AlSb diamondoids are used as building blocks to investigate AlSb nanocrystal properties using density functional theory. Energy differences between the HOMO and LUMO of AlSb diamondoids vary according to confinement theory, along with shape fluctuations. AlSb diamondoids' vibrational force constant reaches 0.82 mDyne/Å, which is less than that of bulk tin. Al-Sb octamantane's vibrational frequencies and reduced masses reach 334.4 cm−1 and 43.5 amu, respectively. Size variations of UV-Vis show that the maximum optical peak moves from 117 nm to nearly 434.4 nm as the size of the AlSb diamondoids and molecules increases. NMR spectra of AlSb diamondoids are analysed as a function of the diamondoids' size. 1H-NMR shielding of AlSb diamondoids shows values that are split, in which Al-H shielding is lower than Sb-H shielding. Natural-bond orbital population analysis shows that the present diamondoids' bonding differs from ideal diamond sp3 hybridization bonding. The bonding for AlSb electronic orbitals at the centre of AlSb octamantane is Al([core]3s0.913p1.744p0.02) Sb([core]5s1.395p4.056p0.01). The electronic occupation depends on the distance between AlSb atoms and the diamondoid's surface. AlSb diamondoids' vibrational longitudinal optical mode is red shifted with respect to the experimental bulk value, which is the case for C and Si.
1. Introduction
Aluminium antimonide is a very important semiconductor, especially within the nanoscale range. The energy gap of bulk AlSb is 1.6 eV [1]. This gap is enlarged as we go to the nanoscale, according to quantum confinement theory. Enlarging the AlSb gap will result in a gap that can cover the visible spectrum (1.65–3.1 eV) and part of the ultraviolet range. Alloying AlSb with other III-V compounds, such as GaSb, InSb and AlAs, can make the gap vary in a wide range of the spectrum for different applications. Current applications of AlSb include electronics [2, 3], optoelectronics [4] and solar cells [5].
Diamondoid structures are hydrogenated carbon cage clusters that are very close to bulk diamond. They are found to be stable, at least theoretically, for all group IV elements, zincblende and many other cage-like structures [6, 7]. Lab preparation of these structures is also in progress [6]. The present trend of size reduction, lightweight and tight structures, etc., are all preferred properties for applications in molecular electronics. These properties are applicable for diamondoids of carbon and all other elements, and for compounds that can form such structures, including the present work suggesting the use of AlSb diamondoids. We aim to prove that we can achieve bulk AlSb properties through increasing the AlSb diamondoids' size. Hydrogen is an essential part of the structure of diamondoids; without it, their structure either becomes unstable or deviates significantly from the sp3 orbital hybridization bonding that is a distinctive characteristic of diamond and diamondoids. Bare group IV clusters [8, 9] have either sp or sp2 orbital bonding on their surface and a smaller gap, which should also be the case of bare III-V compounds such as AlSb diamondoids.
2. Theory
In order to choose between different theories of density functional theory (DFT) and the available basis functions, we performed the following benchmark test. Experimental data are mainly available for carbon diamondoids. The experimental energy gap of carbon adamantane is 6.492 eV [10]. As we increased the size of the diamondoids, this energy gap decreased. Heavy elements like Sb have limited basis state choices. Our choice was the 3–21G basis, since it is the best available choice for Sb. The 3–21G basis is mostly adequate for ground-state calculations, such as geometry, binding energy, charge density and vibrational properties, including transitions to ground state. Performing electronic structure calculations of carbon adamantane using the above basis with different theories, such as PBE, LSDA, BLYP and B3LYP, we got the following energy gaps 7.95, 7.75, 7.95 and 9.80, respectively [11]. With the exception of the B3LYP method, all other results are close to each other and are approximately 1.4 eV higher than the experiment result. The B3LYP is nearly 3.3 eV higher than that of the experiment. On the other hand, DFT methods are known to underestimate the value of bulk diamond [12]. The value of the gap using LSDA theory and the advanced (cc-pV(T +d)Z) basis set is 6.472 eV [11], in comparison with the experimental value of C-adamantane 6.492 eV. The B3LYP method result for the (cc-pV(T+d)Z) basis set is 8.182 eV, which explains why we preferred the LSDA method over other methods.
To calculate the electronic structure of AlSb diamondoids and molecules, all-electron density functional theory at the level of local spin density approximation (LSDA) with 3–21G basis was used. We started with two small nondiamondoid molecules, i.e., AlSbH6 and AlSb cyclohexane (Al3Sb3H12). AlSbH6 is the smallest molecule that contains an Al-Sb bond, while Al3Sb3H12 are the faces of the blocks that are assembled to construct AlSb diamondoids. Diamondoids are named with reference to the number of cages in their structures. AlSb diamondoids investigated in the present work include AlSb diamantane (Al7Sb7H20), AlSb tetramantane (Al11Sb11H28), AlSb hexamantane (Al13Sb13H30) and AlSb octamantane (Al20Sb20H42). Some of these structures are shown in Fig (1). The last molecule can be considered a nanocrystal since at least one of its dimensions is greater than 1 nm.

AlSb diamantane-, tetramantane-, hexamantane- and octamantane-optimized atomic sites
The vibrational force constant, reduced masses, NMR and UV-Vis spectra of some of the present AlSb diamondoid molecules and nanocrystals were also calculated. Heavier AlSb diamondoids' spectroscopic calculations require computational resources that are not available in our system. The frequencies of vibrational spectra are corrected using the 0.984 scale factor that is usually used with the present theory and basis (LSDA/3-21G) [11]. The optimized geometries are also used to calculate 1H-NMR shielding. The UV-Vis is calculated using excited state energy calculation including up to 40 states with the time-dependent density functional theory (TD-DFT) method. Some molecules, like AlSbH6 have 28 empty states only in the present theory, so we actually calculated all the possible transitions in this molecule. Natural bond orbital (NBO) population analysis was performed to study the kind of hybridized molecular orbitals that formed in the present diamondoids. All calculations were performed using the Gaussian 09 program [13].
3. Results and Discussion
Fig (2) shows the energy gap variation with the total number of Al and Sb atoms as predicted by the present theory. The highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) are also shown in Fig (2). The dashed line represents the bulk gap experimental limit of these diamondoids at 1.6 eV [1]. The mostly descending values of the gap are in agreement with the confinement theory's results. Fluctuations are to be expected because of the non-spherical shape of these diamondoids [14, 15]. In general, isomers that have nearly equiaxed molecules or nanocrystals were investigated in the present work. The deviation at 26 AlSb atoms indicates a shape change between AlSb tetramantane and AlSb hexamantane. The change in the HOMO level is slightly larger than the LUMO level that contributes to the change of the energy gap.
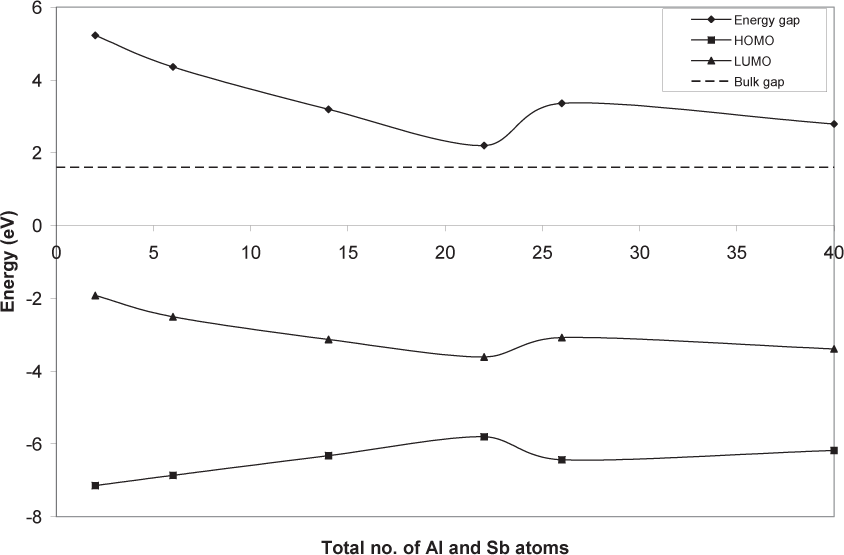
Energy gap variation with the number of AlSb atoms as predicted by the present theory. The highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) are shown. The dashed line represents the experimental bulk gap limit at 1.6 eV.
Figs (3) and (4) show the evolution of AlSb diamondoids' vibrational force constant and reduced mass for AlSbH6 and AlSb octamantane, respectively, as a function of vibrational frequency. These figures can be divided into two regions. The first one is approximately from 0 to 340 cm−1. In this region, both the force constant and reduced mass are oscillating. The highest force constant in this region starts with the value 0.24 mDyne/Å for AlSbH6 and ends with 0.82 mDyne/Å for AlSb octamantane. The last value is less than that of bulk tin (1.3 mDyne/Å) [16] and that of bulk silicon (2.4 mDyne/Å) [17] (Sn and Si are the closest elements to Sb and Al in terms of atomic mass). The highest force constant mode (HFCM) in this region is the longitudinal optical mode (LO) [18, 19]. The highest reduced mass also increases from 10.8 amu for AlSbH6 to 43.53 amu for octamantane. The combined reduced mass of Al and Sb atoms is approximately 22.1 amu. The combined reduced masses of two Al or two Sb atoms are 13.5 and 60.8 amu, respectively. This proves that some vibrations are due to the movement of mainly Al atoms only, or Sb atoms only. For a material that is composed of two atoms that have a high difference in their masses, the limits of vibrational frequency at the Brillouin zone boundary are approximately given by [20]:
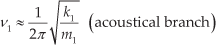


Variation of the vibrational force constants in (mDyne/Å) as a function of frequency for AlSbH6 and AlSb octamantane. The dashed line represents the experimental bulk value of the LO mode's frequency [20].

Variation of the vibrationally reduced masses in atomic mass units (amu) as a function of frequency for AlSbH6 and AlSb octamantane. The dashed line represents the experimental bulk value of the LO mode's frequency [20].
where m1 and m2 are the light and heavy atoms that are Al and Sb atoms, respectively, in the present compound and k is the force constant. The frequency of the HFCM increases from 191.4 cm−1 for AlSbH6 to 334.4 cm−1 for AlSb octamantane. The latter value is lower (red shifted) than bulk AlSb (337 cm−1) [21, 22]. The LO mode frequencies are 316.4, 330.1, 329.2 and 334.4 cm−1 for the diamondoid clusters AlSb diamantane, tetramantane, hexamantane and octamantane, respectively, which are all red shifted with respect to bulk 337 cm−1.
The second part of Figs (3) and (4) goes approximately from 400 to 1900 cm−1. This part has a nearly constant hydrogen-reduced mass, which is approximately equal to 1 amu. This part includes hydrogen scissor vibrations at nearly 627 and 742 cm−1 for H atoms that are connected to Al and Sb surface atoms, respectively. Symmetric and asymmetric vibrations are at the end of the spectrum, in the range 1786–1866 cm−1.
UV-Vis spectra of some of the investigated molecules and diamondoids in the present work (up to AlSb tetramantane) are shown in Fig (5). The first two spectra are for AlSbH6 and AlSb cyclohexane molecules, respectively. These molecules have higher UV-Vis spectra in comparison with diamondoids' molecules that are presented in the same figure, i.e., AlSb diamantane and tetramantane. The reason for this difference is the existence of high ionic charges on AlSbH6 and AlSb cyclohexane, induced by the high number of H atoms with respect to the number of Al and Sb atoms. This shows that diamondoid molecules have reduced relative UV-Vis intensity with respect to non-diamondoids, as in Fig (5). The peak maximum in UV-Vis spectra moves from 113 nm for AlSbH6 to 440 nm for AlSb tetramantane. The later variation of values is toward the cut-off energy at 775 nm, as implied by the value of the bulk energy gap 1.6 eV [1]. An accuracy problem prohibited us from adding AlSb hexamantane and octamantane UV-Vis spectra to Fig 5.

Variation of UV-Vis spectra as a function of excitation energy for hydrogenated AlSb molecules and diamondoids. The dashed line represents the bulk value of cut-off energy in AlSb, as implied by the energy gap value [1].
Electronic spin produces a magnetic field (shielding) that is opposite to the magnetic field produced by the nucleus. The Gauge-Independent Atomic Orbital (GIAO) method was used in the present NMR calculations. Size variations of 1H-NMR shielding of AlSb diamondoids are shown in Fig (6). The split of upper and lower 1H-NMR lines are shown in this figure. Because of symmetry, the first point in Fig (6) that corresponds to AlSbH6 shows that minimum and maximum 1H-NMR spectra lines almost coincide. The second line of points corresponds to AlSb cyclohexane. These two molecules (non-diamondoids) and other diamondoids show shielding values that are split, in which Al-H shielding is lower than Sb-H shielding. This clear behaviour is due to the different electronic environments around the two atoms Al and Sb. Al usually loses some electronic charge to surrounding Sb atoms and also to H surface atoms. Sb has the opposite behaviour, as we can see from NBO analysis below.

Size variations in 1H-NMR shielding of AlSb diamondoids and molecules
NBO analysis of hydrogenated AlSb diamondoids and molecules can be used to reveal the kind of bonding between the constituting atoms. The present AlSb diamondoids differ from bulk-diamond ideal sp3 hybridization bonding. The bonding is in the range of Al([core]3s0.913p1.744p0.02) Sb([core]5s1.395p4.056p0.01) at the centre of AlSb octamantane to Al([core]3s0.953p1.664p0.01) Sb([core]5s1.475p3.716p0.01) at the surface. Surface Al atoms lost some of their electronic charge to Sb and surface H atoms, as is obvious from the sum of the orbital occupation at the above centre and surface configurations, in which the total electronic Al occupations are 2.67 and 2.62 electrons (instead of three electrons in bare Al atoms), respectively. The reverse is true for Sb atoms. The s orbital occupation is greater than one for the heavier Sb atom, which is the expected trend of occupation for heavy atoms [23]. This can be compared to diamond (carbon) atom occupation in which sp3 bonding occurs. Heavier group IV elements, such as Ge and Sn, are similar to Sb and tend to hybridize orbitals towards free atom hybridization s2p2 [23].
Fig (7) shows the structural parameters of AlSb octamantane, which include bond length and tetrahedral angles. The experimental value of the Al-Sb bond length of 2.65 Å [1] is indicated in the first figure (7a) as a dashed line. This line is nearly at the middle of the Al-Sb bond distribution. The average experimental value of the Al-H bond length is 1.62

Structural parameters of AlSb octamantane, which include (a) bond length and (b) tetrahedral angles. The experimental value of the Al-Sb bond length [1] is indicated in the first figure (a) as a dashed line. The typical value of tetrahedral angles (109.47 degrees [15]) in diamond and zincblende structures is indicated in the second figure (b).
Fig (8) shows the calculated binding energy per atom for AlSb diamondoids and molecules, as a function of the number of atoms compared with experimental bulk value at 6.61 eV [24]. For small molecules, the binding energy exceeds that of the bulk, due to the effect of H strong bonds at the surface. As the number of Al and Sb atoms increases, this effect diminishes and the binding energy stabilizes at a certain value slightly lower than the experimental value. In the procedure of calculating the optimum positions of atoms, both the forces and the displacement of all atoms are reduced below a certain threshold value, which practically means that the atoms are influenced by almost no forces and are fixed in their positions. Maximum and RMS forces are less than 0.00045 and 0.0003 Hartrees/Bohr, respectively. Maximum and RMS displacements are less than 0.0018 and 0.0012 Å, respectively. Adding these optimization criteria to the calculated binding energy in Fig (8) ensures the stability of these structures. Finally, a wide range of applications of carbon diamondoids are recognized [25-27], which include biomedicine, the material sciences, nanotechnology optoelectronics and petroleum science. These applications will surely include other kind of diamondoids, including the presently used one.

Binding energy per atom as a function of number of AlSb atoms. The dashed line represents the experimental bulk value of the binding energy [24].
4. Conclusions
AlSb diamondoids have properties that distinguish them from other AlSb molecules and bulk. These properties include their relatively tight structure, stability, reduced relative UV-Vis intensity with respect to non-diamondoids and split 1H-NMR shielding, in which Al-H shielding is lower than Sb-H shielding. These properties were visualized through the calculated vibrational-reduced masses, force constants, UV-Vis spectra and 1H-NMR lines. Most of the calculated properties in the present work tend to converge to experimental bulk properties, if available. This convergence was sometimes approached from values higher than bulk, such as some vibrational frequencies and binding energy per atom, due to the existence of hydrogen surface atoms. AlSb diamondoid electronic and structural properties were proved to be compatible with experimental values, such as energy gap, binding energy, bond lengths and tetrahedral angles. AlSb diamondoids' vibrational LO mode was red shifted with respect to the bulk experimental value, which was also the case for C and Si.