
Abstract
Nuclear magnetic resonance measurements confirm that, as expected, the mer isomer of tris(β-diketonato) cobalt(III) complexes is more stable (mer isomers are more stable because they are less affected by steric hindrance and they have lower dipoles). An excess of the mer isomer or only the mer isomer is experimentally observed. The existence of both fac and mer isomers is predicted by density functional theory calculations. At room temperature, the nuclear magnetic resonance peaks of phenyl-containing mer complexes are broad due to the intermediate rotation of bulky phenyl groups resulting in only the average orientation of the non-equivalent protons in the mer complexes. However, upon heating to 55 °C (thus speeding up the rotation) or cooling to −50 °C (slowing down the rotation), this broad peak is resolved into three sets of carbon peaks appearing in the 13C NMR spectrum, as well as splitting of the 1H NMR peaks for the non-equivalent protons in the mer isomer. In solid-state 13C NMR the aromatic rings do not rotate, resulting in the splitting of all the 13C NMR peaks. Two sets of well-defined peaks are observed for the fluorine-containing compound [Co(CF3COCHCOCPh)3] in both the 13C and 19F NMR spectra, indicating that both the fac and mer isomers are present with a ratio of fac:mer = 0.1:1. The X-ray photoelectron spectroscopy measured binding energy of the Co 2p3/2 photoelectron lines are influenced by the subtle electronic (electron-donating or electron-withdrawing) properties of the R groups on the β-diketonato ligands. Various relationships are established between the binding energy of Co 2p3/2 and several physical and density functional theory–calculated properties, confirming good electronic communication (the measurable electronic effect that a molecular fragment has on the physical properties of another molecular fragment in the molecule) of the substituent groups through the pseudo aromatic system of the β-diketonato backbones to the metal.

Peak resolution in the NMR is dependent on the rotation speed of the phenyl rings, which in turn is dependent on the temperature; however, the experimental fac/mer isomer ratio is independent of temperature of Co(β-diketonato)3.
Highlights
Density functional theory (DFT) calculations show that both fac and mer isomers are possible for Co(β-diketonato)3, as indeed been experimentally observed.
At room temperature, nuclear magnetic resonance (NMR) spectra revealed one broad peak representing the average aromatic entities of the non-equivalent protons of the mer isomer.
High- and low-temperature NMR resulted in three well-defined peaks for the aromatic entities of the mer isomer.
From relationship between binding energy (BE) of Co 2p3/2 and other physical properties, BE for unknown compounds can be predicted.
Introduction
β-Diketones (the most well-known type is acetylacetone (Hacac)) can easily coordinate to many metals forming square planar,1,2 octahedral,3–5 or eight-coordinated complexes.6,7 This property of β-diketones is applied in the solvent extraction of metals, for example, cobalt(II) can be extracted almost completely (95%–99.5% by a single extraction) under the correct experimental conditions, forming the very stable tris(acetylacetonate)cobalt(III) complex.
8
Many metal-β-diketonato complexes have applications in industry, both as homogeneous and heterogeneous catalysts. For example, [Co(acac)3] is used as a homogeneous catalyst in many reactions, such as cycloaddition, cyclization, co-dimerization and olefin coupling reactions, organometallic coupling and addition reactions, radical addition to olefins, and oxidation reactions.
9
Different tris(β-diketonato)cobalt(III) complexes were tested as catalysts for the hydration of olefins. The yield was dependent on the type of tris(β-diketonato)cobalt(III) complex used, as well as the type of olefin substrate catalyzed, though Co(III)-containing 1,3-substituted β-diketones in many cases performed better than [Co(acac)3].
10
The different electron-donating and electron-withdrawing properties of the substituents R1 and R2 on the β-diketonato ligand, (R1COCHCOR2)−, donate or withdraw electron density via the pseudo aromatic backbone to or from the metal it is coordinated to, thereby influencing the reactivity of the metal. The influence of the electron-donating and electron-withdrawing properties of the substituents R1 and R2 on the electron-richness of the metal can inter alia be measured by redox potential of the metal, which also relates to various experimental properties (pKa,
8
X-ray photoelectron spectroscopy (XPS) binding energies,11,12 reduction potential of the free β-diketone13,14), empirical parameters (Hammett constant,15,16 Lever parameters,
17
Gordy scale group electronegativities
18
), and theoretically calculated energies (frontier orbital energies,19,20 electron affinity (EA), ionization potential (IP), the Mulliken electronegativity (χ),21,22 the global electrophilicity index (ω)
23
) of the substituents R1 and R2 of the β-diketone R1COCH2COR2 or the metal-β-diketonato complex. To our knowledge, the binding energies (BE) of the core Co 2p electrons, as measured by XPS, of only one tris(β-diketonato)cobalt(III) complex, the tris(acac)cobalt(III) complex, has been reported.
24
In this contribution, we determine the electronic influence of the substituents R1 and R2 on cobalt(III) in tris(β-diketonato)cobalt(III) complexes, [Co(R1COCHCOR2)3], by nuclear magnetic resonance (NMR) and XPS spectroscopy, combined with a theoretical density functional theory (DFT) study of five tris(β-diketonato)cobalt(III) complexes containing different β-diketonato ligands, (R1COCHCOR2)− with R1, R2 = CH3, CH3 (

The tris(β-diketonato)cobalt(III) complexes in this study: [Co(acac)3] (
Results and discussion
The tris(β-diketonato)cobalt(III) complexes [Co(acac)3] (

(a) The Λ and Δ stereoisomers, and (b) the fac and mer isomers of [Co(β-diketonato)3] complexes, containing an unsymmetrical β-diketonato ligand (R1COCHCOR2)– with R1 ≠ R2 and (c) an illustration of the statistical fac: mer = 1:3 distribution. The blue arrows in (a) indicate the right-handed propeller twist of the R1 and R2 groups of the bidentate ligand for the Δ stereoisomer, whereas for the Λ stereoisomer the left-handed propeller twist of the R1 and R2 groups is stipulated. The pink arrows in (c) indicate the spacial orientation of the R1 groups on different bidentate ligands which results in the statistical 3 (mer):1 (fac) ratio. Note mer-1, mer-2, and mer-3 are identical and only illustrate the statistical population of the arrangement of the ligands around Co. Thus, according to statistics, each fac molecule forms 3 mer molecules.
DFT study
DFT calculations using the B3LYP/6-311G(d,p)/Lanl2dz(Co) theory level showed that the DFT-optimized Λ and Δ isomers of each molecule have the same bond lengths and angles and are equi-energetic. The DFT-calculated free energy of the fac and mer isomers depends on their substituents and the temperature. It was found that the mer isomers are more stable for

Temperature-dependent populations of fac and mer isomers of
The DFT calculations, in agreement with experimental observations, thus show that both fac and mer isomers are experimentally possible. In addition, it has been experimentally observed that the yield of the minor fac isomer of
NMR studies
Using NMR, it is possible to distinguish between a fac and a mer isomer for
The liquid-state 1H and 13C NMR spectra of compounds containing symmetrically substituted β-diketonato ligands
Compound

1H NMR spectrum of

19F NMR spectrum of

13C NMR spectrum of
The 13C NMR spectrum of
For

Liquid state at 25 °C (bottom) and solid-state (top) 13C NMR spectra of
For both
Contrasting
The 19F NMR spectrum of compound
Thus, the NMR results of this study show that the fac:mer ratio of a sample of compounds
XPS study
XPS studies have been undertaken pertaining to the study of the electronic structure of
When a sample is exposed to the atmosphere, large quantities of adventitious carbon deposit on the sample. The main simulated C 1s photoelectron line of the adventitious carbon (284.8 eV) is used as a reference to correct the position of the photoelectron lines of the elements due to any charging that might have occurred during the measurement.
Figure 8 shows the detailed comparative XPS spectra of the Co 2p3/2 photoelectron lines of compounds

Detailed XPS spectra of the Co 2p3/2 region for
The accumulative Gordy group electronegativity of the R groups of the β-diketonato ligands, ΣχR, the accumulative Hammett constants (ΣσR), the pKa of the β-diketonato ligand, the DFT-calculated electrophilicity index (ω in eV), the electrochemical potential (μ in eV) and the Mulliken electronegativity (χ in eV), the Lever constants (LE) as well as E(HOMO) (in eV) and E(LUMO) (in eV), and the DFT-calculated energy of the Co 2p orbital energy, E(Co 2p) (eV) of

DFT: density functional theory; E(HOMO): energy of the highest occupied molecular orbital; E(LUMO): energy of the lowest unoccupied molecular orbital; BE: binding energy; FWHM: full width at half maximum.
The XPS measured binding energy of the main Co 2p3/2 photoelectron line (in eV), the full width at half maximum (FWHM in eV) of the main Co 2p3/2 photoelectron line, the spin-orbit splitting between the Co 2p1/2 and Co 2p3/2 photoelectron lines (ΔBE1 in eV), the sum of the intensity ratio of all the satellite structures and the intensity of the main Co 2p3/2 photoelectron lines (IΣ = (ICo 2p3/2 sat1 + ICo 2p3/2 sat2 + ICo 2p3/2 sat3)/(ICo 2p3/2 main)), as well as the binding energy of the F 1s photoelectron line (in eV) of
Sum of the R group electronegativities, ΣχR. χCF3 = 3.01, χCH3 = 2.34, and χC6H5 = 2.21.2,39 By way of example, for
pKa from Starý. 8
LE from Lever. 17
DFT-calculated ωR, μ, χ (χ = −µ (μ is the electrochemical potential), E(Co 2p), E(HOMO), and E(LUMO) data from this study. DFT-calculated values for the fac and mer isomers were the same within 0.02 V, thus the values obtained for the fac isomers are reported here.
The subtle influence of the electronic (electron-donating or electron-withdrawing) properties of the R groups on the β-diketonato ligands on the chemical environment of the Co center can be graphically displayed from the plot of the binding energies of the main Co 2p3/2 photoelectron lines (BE Co 2p3/2 main) of
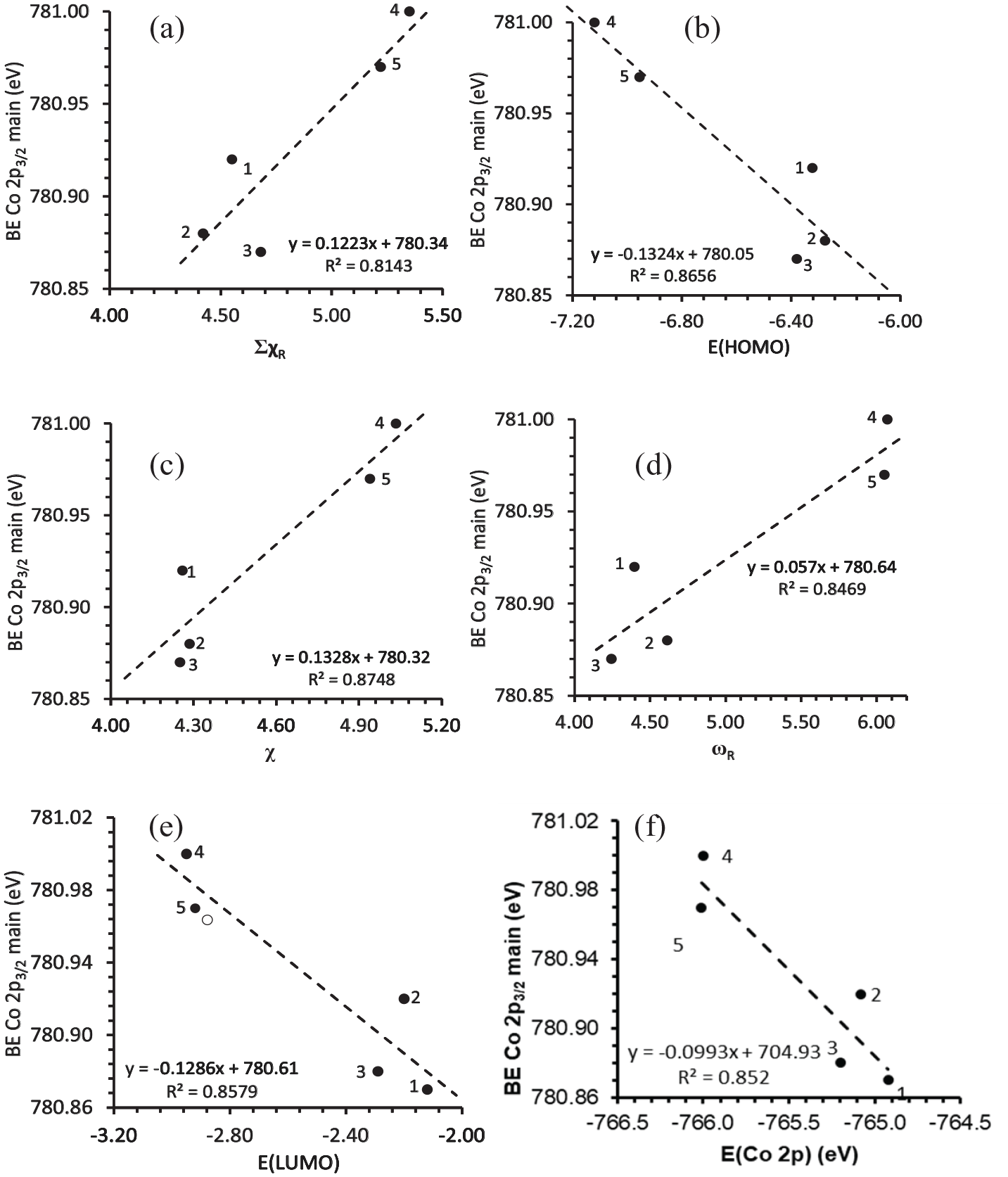
The relationship between the binding energy of the main Co 2p3/2 photoelectron line (BE Co 2p3/2 main) and (a) the sum of Gordy scale R group electronegativities of the β-diketonato ligands, ΣχR, of
The energy of the highest occupied molecular orbital (E(HOMO)) and BE Co 2p3/2 main are inversely related to each other (Figure 9(b)). The lower the energy of the HOMO implies that more energy is required to remove an electron occupying the HOMO, that is, to ionize the complex.20,23 Thus, the more difficult it is to ionize a complex (to remove an electron from a lower energy Co d HOMO), the more difficult it is to emit a photoelectron from the Co 2p orbital (as indicated by the higher BE Co 2p3/2 main) on this metal ion. Additional relationships were established between BE Cu 2p3/2 main and the DFT-calculated properties, that is, the calculated Mulliken electronegativity (χ, a measure of the tendency of an atom or molecule to attract electrons)
21
and the global electrophilicity index (ω, a measure of the electrophilic power of atoms and molecules)
23
confirm the dependence of the BE Co 2p3/2 main on the initial state of (
Further relationships between BE Co 2p3/2 main and the pKa of the β-diketonato ligand, the Hammett constants (Σσ), the DFT-calculated electrochemical potential (μ), and the Lever constants (LE) can be found in the Supporting Information. The equations and R2 values, obtained from the relationships involving BE Co 2p3/2 main, are summarized in Table 2. Using these relationships involving BE Co 2p3/2 main, it is potentially possible to predict the BE of a related complex, for example, Co(hfaa)3 (hfaa = CF3COCHCOCF3) as 781.09 with a mean absolute deviation (MAD) of 0.02.
Relationships obtained between XPS measured binding energies of the main Co 2p3/2 photoelectron lines (in eV) and the Gordy group electronegativities of the R groups of the β-diketonato ligands, ΣχR, the accumulative Hammett constants (ΣσR), the pKa of the β-diketonato ligand, the DFT-calculated electrophilicity index (ω in eV), the electrochemical potential (μ in eV) and the Mulliken electronegativity (χ in eV), the Lever constants (LE), as well as the E(HOMO) (in eV) and E(LUMO) (in eV).

XPS: X-ray photoelectron spectroscopy; DFT: density functional theory; E(HOMO): energy of the highest occupied molecular orbital; E(LUMO): energy of the lowest unoccupied molecular orbital; BE: binding energy; MAD: mean absolute deviation.
Predicted BE of the Co 2p3/2 main photoelectron for Co(hfaa)3 using the tabulated equations.
To further validate the predicted BE Co 2p3/2 main for Co(hfaa)3 (hfaa = CF3COCHCOCF3) as 781.09 (Table 2), the Co 2p orbital energies, that are related to XPS IPs, were determined using DFT, and the values are given in Table 1. The relationship between the XPS measured binding energy of the main Co 2p3/2 photoelectron line and the DFT-calculated Co 2p orbital energies, as shown in Figure 9(f), is
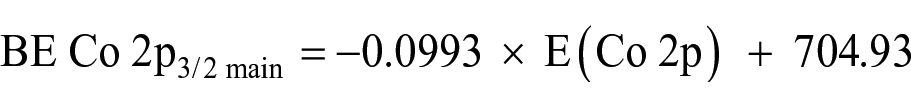
Using this equation and the calculated Co 2p orbital energy of −767.11 for Co(hfaa)3, a value of 781.10 eV is obtained, being inherently the same value as that predicted from the relationships shown in Table 2.
A spin-orbit splitting (ΔBEmain = BECo2p1/2 main − BECo2p3/2 main) of 15.3–15.6 eV was observed, depending on the electronic properties of the R group on the β-diketonato ligands (Figure 10(a)). This peak separation (ΔBEmain) can be used to quantify the delocalization of the valence electrons38,40 and their weakened interaction with the core. Since a smaller separation (ΔBEmain) is associated with a higher degree of delocalization of the unpaired electrons,36,41 it is implied that Co compounds with more electron-donating groups (lower ΣχR) display smaller ΔBEmain values, and accordingly have increased delocalization of the 3d valence electrons, see Figure 10(a). The average ΔBEmain of ca. 15.4 eV is characteristic of diamagnetic Co(III) compounds. 38

(a) The relationship between the spin-orbit splitting of the main Co 2p3/2 and Co 2p1/2 photoelectron lines (ΔBEmain = BE Co 2p1/2 – BE Co 2p3/2) and ΣχR, and (b) the relationship between FWHM of the main Co 2p3/2 photoelectron line and the sum of the β-diketonato ligand Gordy scale R group electronegativities, ΣχR, of
The uncertainty principle predicts that a lifetime of the core-hole created by photoemission will result in a wider full width at half maximum (FWHM).
42
The FHWM can thus be used as an indication of the lifetime of a core-hole (since the time of measurement for all the samples were the same). From the trend shown in Figure 10(b), the lifetime of the core-hole created by photoemission is linearly dependent on ΣχR. Thus, the more the electron density withdrawn from the Co(III) center (e.g.
Despite reports stating that diamagnetic Co(III) displays no to weak satellite structures,
38
and that the XPS data of Co(acac)3 reported in 1975 gives weak satellites,
24
the Co(β-diketonato)3 complexes

XPS of the Co 2p photoelectron lines of
Conclusion
An insight into how the electronic properties of the R groups on the β-diketonato ligands influence its communication with the central cobalt atom may be obtained by critically evaluating the relationships between the XPS measured binding energy of the Co 2p3/2 photoelectron lines and the sum of Gordy scale R group electronegativities of the β-diketonato ligands, the E(HOMO), E(LUMO), the Mulliken electronegativities, and the global electrophilicity indices. Using these relationships as well as the DFT-calculated Co 2p orbital energies, the binding energy of an unknown tris(β-diketonato) colbalt(III) can be predicted with high accuracy (MAD of 0.02). In addition, from the NMR results, it can be concluded that % of fac/mer isomers in a sample is not influenced by temperature or time when dissolved in solution (thus no dynamic equilibrium exists). It is therefore assumed that the % of fac/mer isomers of a sample is a consequence of the experimental conditions.
Experimental
Materials and methods
Reagents were obtained from Merck and Sigma-Aldrich. Solid reagents employed in preparations were used directly, without further purification. Liquid reactants and solvents were dried and distilled prior to use. Melting points (m.p.) were determined using an Olympus BX51 system microscope, assembled on top of a Linkam THMS600 stage, and connected to a Linkam TMS94 temperature programmer.
Synthesis
Complexes

Addition of H2O2 to the ligand suspension caused vigorous frothing; therefore, the addition of H2O2 was done very slowly. The reaction mixture turned dark green as coordination to the oxidized Co(III) occurred. After 40 min, the reaction mixture was cooled on ice and the obtained green precipitate was filtered and washed with excess cold water and then dried in a desiccator at room temperature. The complexes do not dissolve in water; they are, however, slightly soluble in acetone and soluble in methanol, acetonitrile, and most halogenated organic (chloroform) solvents. The different melting points reported in the literature are due to the different crystalline phases and different melting points for the fac and mer isomers. 30
Characterization data for [Co(CH3COCHCOCH3)3], (
Characterization data for [Co(C6H5COCHCOCH3)3], (
Characterization data for [Co(C6H5COCHCOC6H5)3], (
Characterization data for [Co(CH3COCHCOCF3)3], (
Characterization data for [Co(C6H5COCHCOCF3)3], (
Spectroscopic analysis
Nuclear magnetic resonance spectroscopy
The liquid-state 1H, 13C, and 19F NMR spectra were recorded at 25.0 °C on a 600 MHz Avance II Bruker spectrometer operating at 600.28, 150.95, and 564.83 MHz for 1H, 13C, and 19F, respectively. Deuterated chloroform was used as the solvent. The chemical shifts (δ) are reported in parts per million (ppm) and the spectra are referenced relative to the Me4Si internal standard at 0 ppm for 1H and 13C spectra. Coupling constants (J) are reported in Hz. The solid-state NMR spectra were collected on a 400 MHz Bruker Avance III spectrometer equipped with a 4-mm VTN multinuclear double resonance magic angle spinning probe, operating at 25.0 °C. The 13C NMR spectra were recorded at 100.61 MHz, using the cross polarization magic angle spinning (CP/MAS) technique. A rotating speed of 14,000 Hz was used with a contact time of 2 ms, a recycle delay of 5 s, and an acquisition time of 33.9 ms. The sample was packed in a 4-mm zirconia rotor. 19F NMR chemical shifts are given relative to hexafluorobenzene in CDCl3 at δ = −164.9 ppm (external reference).
X-ray photoelectron spectroscopy
XPS data were recorded on a PHI 5000 Versaprobe system with a monochromatic Al K X-ray source. Spectra were obtained using the aluminum anode (Al Kα = 1486.6 eV) operating at 50 μm, 12.5 W, and 15 kV energy (97 X-ray beam). The survey scans were recorded at a constant pass energy of 187.85 eV and region scans at pass energies ranging from 29 to 90 eV with the analyzer resolution ⩽0.5 eV. The background pressure was ca. 2 × 10−8 mbar. Spectra have been corrected to the main line of the adventitious carbon 1s spectrum, which was set to 284.8 eV. The XPS data were analyzed utilizing Multipak version 8.2c computer software, 53 using Gaussian–Lorentz fits (the Gaussian/Lorentz ratios were always >95%).
Matrix-assisted laser desorption ionization time-of-flight
MALDI-TOF spectra were collected using a Bruker Microflex LRF20 in positive mode with 2-[(2E)-3-(4-tert-butylphenyl)-2-methylprop-2-enylidene]malononitrile (DCTB) as the matrix. A minimum laser power 337 nm was required to observe signals.
Density functional theory calculations
DFT calculations were performed using the B3LYP functional, as implemented in the Gaussian 16 package, 54 using the triple-ζ basis set 6-311G(d, p) for all non-metal atoms, and the Lanl2dz basis set 55 that corresponds to the Los Alamos ECP plus DZ was used for both the metal electronic core and valence electrons. The B3LYP exchange-correlation functional includes 20% Hartree-Fock exchange and GGA corrections (Becke 88 exchange functional and the Lee, Yang and Parr correlation functional) in addition to LDA (VWN local-density approximation) electron-electron and electron-nuclei energy. 56 B3LYP is well established in the literature and is on average the best choice for most systems, especially small organic systems. 57 All optimized structures have been verified as minima by performing frequency calculations in order to ensure that no imaginary frequency was present. Calculations were done in chloroform implicit solvent, since NMR spectra were obtained in chloroform solution. For solvent calculations, the integral equation formalism polarizable continuum model (IEFPCM) of solvation to describe the dielectric continuum medium was used.58,59 The input coordinates for the compounds were constructed using Chemcraft. 60 The ratio of the relative population of the fac and mer isomers was determined by the Boltzmann equation at temperature T as indicated in Figure 3

where ni is the number of molecules with free energy Gi (fac or mer in this case), with Boltzmann’s constant, k = 1.38066 × 1023 J K−1.
DFT-calculated Mulliken electronegativity (χ), the electronic chemical potential (μ), and the global electrophilicity index (ω) were calculated as described in our previous studies, 61 namely: the EA and IP of each of the compounds were determined from the frontier orbital energies (highest occupied molecular orbital = HOMO and lowest unoccupied molecular orbital = LUMO), according to Koopman’s theorem20,23

The Mulliken electronegativity (χ)21,22 and the global electrophilicity index (ω), defined by Parr and coworkers as the electrophilic power of atoms and molecules, 23 were calculated for each compound, by application of the following formulae

where μ = −(IP + EA)/2 is the electronic chemical potential (the same as −χ = μ) 62 and η = (IP − EA) is the chemical hardness63,64 of the ground state of the atoms and molecules.
The energies of the Co 2p orbital of the molecules are determined by optimizing the molecules using the ADF program system,65,66 and the OLYP basis set,67,68 corrected for dispersion by Grimme’s D3 correction, 69 with an all-electron TZ2P basis set.
Supplemental Material
sj-docx-1-chl-10.1177_17475198231184788 – Supplemental material for Spectroscopic and DFT study of tris(β-diketonato)cobalt(III) complexes
Supplemental material, sj-docx-1-chl-10.1177_17475198231184788 for Spectroscopic and DFT study of tris(β-diketonato)cobalt(III) complexes by Linette Twigge, Jeanet Conradie and Elizabeth Erasmus in Journal of Chemical Research
Footnotes
Credit author statement
JC: Conceptualization; data curation of DFT; interpretation of DFT; writing, reviewing, and editing.
EE: Data curation of XPS; interpretation of XPS; writing, reviewing, and editing.
LT: Data curation of NMR; interpretation of NMR; writing, reviewing, and editing.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The authors are grateful to the Central Research Fund of the University of the Free State and the National Research Foundation (NRF) in South Africa for financial support (Grant nos 129270 (JC) and 132504 (JC)). The High-Performance Computing facility of the UFS, the CHPC of South Africa (Grant no. CHEM0947), and the Norwegian Supercomputing Program (UNINETT Sigma2, Grant No. NN9684K) are acknowledged for computer time. No ethical clearance was required for this study.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.