
Abstract
The electronic, structural and vibrational properties of gallium phosphide diamondoids and nanocrystals were investigated using density functional theory at PBE/6-31(d) level, which included polarization functions. The energy gap obeyed the quantum confinement size effect with shape fluctuations. The gap converged towards its bulk limit at 2.26 eV. The Ga-P bond lengths of higher diamondoids were found to be distributed around the bulk experimental value at 2.36 Angstrom. Tetrahedral angles were found around the ideal bulk zincblende value at 109.47, degrees while dihedral angles were distributed around the ideal bulk zincblende values at ±60 and ±180 degree. These findings illustrate that diamondoids are a good representative of bulk structure. An analysis of vibrational modes, in terms of reduced masses, force constants and IR intensity, was then performed. The size-related change of certain vibrational frequencies of GaP diamondoids was compared with the experimental bulk. Radial breathing mode frequency began from 187 cm−1 for the smallest molecule GaPH6 and decreased with fluctuations, heading to 0 cm−1 as its bulk limit. Longitudinal optical mode began from 187 cm−1 for the smallest molecule and increased with fluctuations, heading to 376.9 cm−1 (11.3 THz) as its bulk limit. Hydrogen-related vibrations were relatively constant and can therefore be used to identify GaP diamondoids because of their high IR and Raman intensity peaks.
Keywords
1. Introduction
Gallium phosphide is a semiconductor with an indirect band gap [1] in its zincblende structure. This semiconductor has allotropic zincblende (cubic) and wurtzite (hexagonal) phases [2]. Its gap in the visible light range, at 2.26 eV, makes it a natural candidate for use in light emitting diodes (LEDs) [1, 3]. In order to manoeuvre the value of this gap, one has to reduce GaP particle size to the nano-scale. The miniaturization of particle size is compatible with the general present trend in reducing electronics' power consumption, making them more high speed and lightweight, etc. To obtain the electronic band structure of GaP molecules or nanocrystals, one has to choose the shape and type of molecule to be considered. An appreciation of the variation of different properties from the molecular to the nano- and bulk-scales is important in order to understand how to deal with these materials in their nanotechnology applications. Recently, a collection of molecules with an increasing number of atoms and varying sizes, called diamondoids, were found to be the building blocks of nanomaterials [4]. These diamondoids are found in nature in petroleum carbon compounds. The subsequent race to prepare carbon diamondoids in the lab can be perceived from the patents on carbon diamondoids [5]. However, theoretically these compounds should exist for all diamond and zincblende structures, and even for other cage-like compounds, such as carbon, silicon germanium, tin, boron nitride, InSb, etc. [6, 7]. Of these, carbon, silicon, boron nitride and other cage molecules and diamondoids have all been prepared in the lab [8, 9]. The theoretical calculations concerning diamondoids have been very useful in themselves, as they have provided insights into how these blocks are combined to form nanocrystals and eventually bulk. The investigation of size-variable GaP diamondoids (Figs. (1–2)) to reach the properties of GaP nanocrystals and bulk limits will be the subject of the present work.

Colour online) Geometrically optimized GaP-diamantane and GaP-tetramantane

Colour online) Geometrically optimized GaP-hexamantane and GaP-octamantane
Diamondoids are strong, cage-like structures that differ from other molecules in the bonding of their surface atoms by one or two hydrogen atoms. This structure results in the bonding of these surface atoms to the core of the molecule or nanocrystal by at least two bonds, which enhances their stability.
2. Theory
The molecular approach to nanocrystal or bulk properties is an interesting approach. This approach can be used to calculate many properties that are sometimes difficult to calculate using solid-state theories. This is partly due to the ab initio molecular programs that have existed since the 1970s. These programs are continuously developing to reach a higher number of atoms with higher precision; one example is the Gaussian 03 program [10] used in the present work. Solid-state programs are more recent and require higher computational resources to include the physical and chemical properties previously included in molecular approaches. The nano-scale region, which is the intermediate region between the molecular and bulk limits, can be approached from one of two approaches, namely, the bottom-up or top-down methods taken from molecular and solid-state theories respectively. In the present work, we shall build GaP nanocrystals starting from the smallest GaP molecules and diamondoids. These molecules include GaPH6 linear molecules, GaP-cyclohexane (Ga3P3H12), GaP-diamantane (Ga7P7H20), GaP-tetramantane (Ga11P11H28), GaP-hexamantane (Ga13P13H30), GaP-octamantane (Ga20P20H42), and GaP-decamantane (Ga22P22H42). The number of cages of these diamondoids is obvious from the nomenclature of the latter five. The first two molecules (which are not diamondoids) were added to complete the overview and range of energy and vibrational spectra. The largest considered GaP diamondoid is GaP-decamantane, which has the dimensions 1.32 nm, 1.06 nm, and 1.06 nm in the standard orientation rectangular (x,y,z) coordinates. The GaP-decamantane can be considered a small nanocrystal. The initial geometries and symmetries are taken from the original geometries of carbon diamondoids. The sizes of the molecules are then scaled up to match the lattice size of GaP diamondoids before optimization. Hydrogen is the original element that passivates carbon diamondoids. Unlike other elements (such as oxygen), it does not severely modify the structure at the surface.
Density functional theory at the Perdew-Burke-Ernzerhof (PBE) level is used in the present work. A 6–31G(d) basis set that includes polarization functions is sophisticated enough to describe GaP diamondoids' orbitals with sufficient accuracy to match experimental properties, as we shall see in the next section. Vibrational frequencies are multiplied by a scale factor 0.986, associated with the 6–31G(d) basis set, which should reduce statistical mismatch between experimental and theoretical results [11]. Using higher basis sets would result in computational difficulties that might have forced us to exclude some of the heavier diamondoids discussed in the present work.
3. Results and discussion
Fig. (3) shows that the energy gap decreases as a function of the total number of Ga and P atoms through most of the investigated range. The exceptions are between GaP-diamantane and GaP-hexamantane. The reason for this deviation is the effect of the shape of the GaP diamondoids. The diamondoids discussed here have unequal lengths in their three dimensions. Since the shape of these diamondoids is not spherical, the laws of quantum confinement do not apply strictly in the present case [12]. In fact, since the present diamondoids are built from nearly cubic cages, the resultant nanoparticles can be of one-, two- or three-dimensional structures. HOMO and LUMO levels are sometimes used as approximations of ionization energy and electron affinity respectively [13]; in Fig. (3), both HOMO and LUMO energy levels are negative. This shows that neither adding nor removing electrons from GaP diamondoids is energetically favourable. This reflects the high stability and inertness of the present diamondoids.
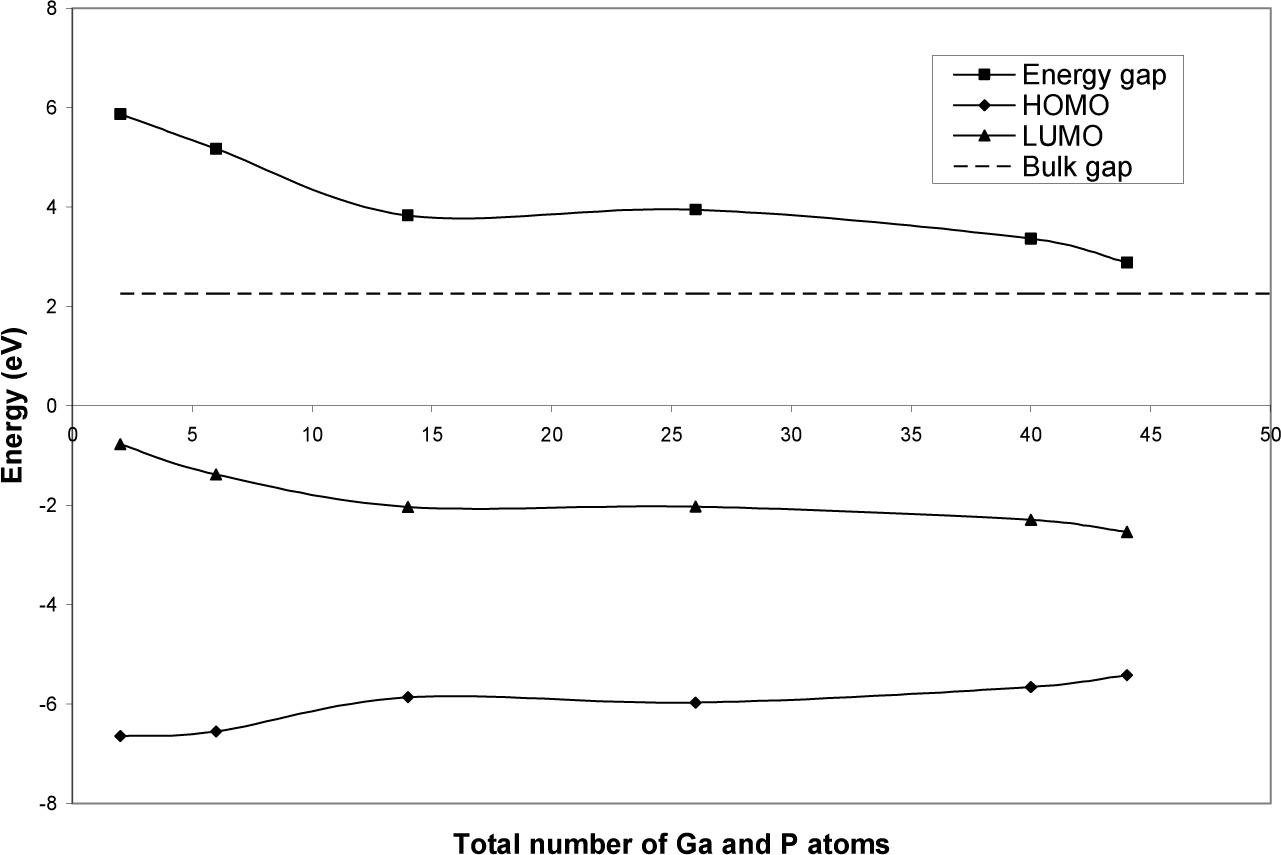
HOMO, LUMO and energy gap of geometrically optimized GaP diamondoids as a function of total number of Ga and P atoms. The dashed line represents the experimental value of the bulk GaP gap [1].
Fig. (4) shows the density of energy states of geometrically optimized GaP-diamantane and GaP-decamantane as a function of energy level. The values of the energy gap (3.83 and 2.88 eV respectively) are blue shifted with respect to the bulk value (2.26 eV) as expected in this range. The near-discrete levels of diamantane turn into the near-continuous band structure of decamantane, which is nearer to the bulk.

Density of energy states of geometrically optimized (a) GaP-diamantane and (b) GaP-decamantane, as a function of energy level. The dotted line represents the Fermi level.
Fig. (5) shows the distribution of bond lengths in GaP-diamantane and GaP-decamantane. The main reason for the nonequivalent bond lengths in our case is the location of atoms either near or far from the surface, and their bonding with either one or two H atoms. The dashed line represents the experimental value of the GaP bulk bond length at 2.36 Å [3]. The figure starts with a sharp, high value for the number of phosphorus-hydrogen bonds. The phosphorus-hydrogen bonds are the shortest bonds in the present molecules. P-H and P-2H refer to the bonding of a phosphorus atom with one or two surface hydrogen atoms respectively. As we can see from Fig. (5), this figure does not distinguish between the two cases P-H and P-2H. On the other hand, Fig. (5) does distinguish between Ga-H and Ga-2H bond lengths for the GaP-decamantane case. The reason for this difference is that P is a nonmetal, while Ga is a metal. Electrons in nonmetals are strongly space-localized with respect to electrons in metals. This results in a difference between Ga-H and Ga-2H bond lengths in GaP-decamantane. The electronic cloud in the singly bonded Ga-H is fed from other delocalized electron clouds on Ga, which results in a longer bond length. This feeding is reduced in the case of Ga-2H, resulting in a shorter bond length. The high number of H atoms relative to Ga atoms in diamantane (nearly three times as many) suppresses this effect. Interestingly, all Ga-P bond lengths are higher than the experimental value in the case of GaP-diamantane (dashed line in Fig. (5a)). The distribution of GaP-decamantane is nearly centred on the experimental value (dashed line in Fig. (5b)).
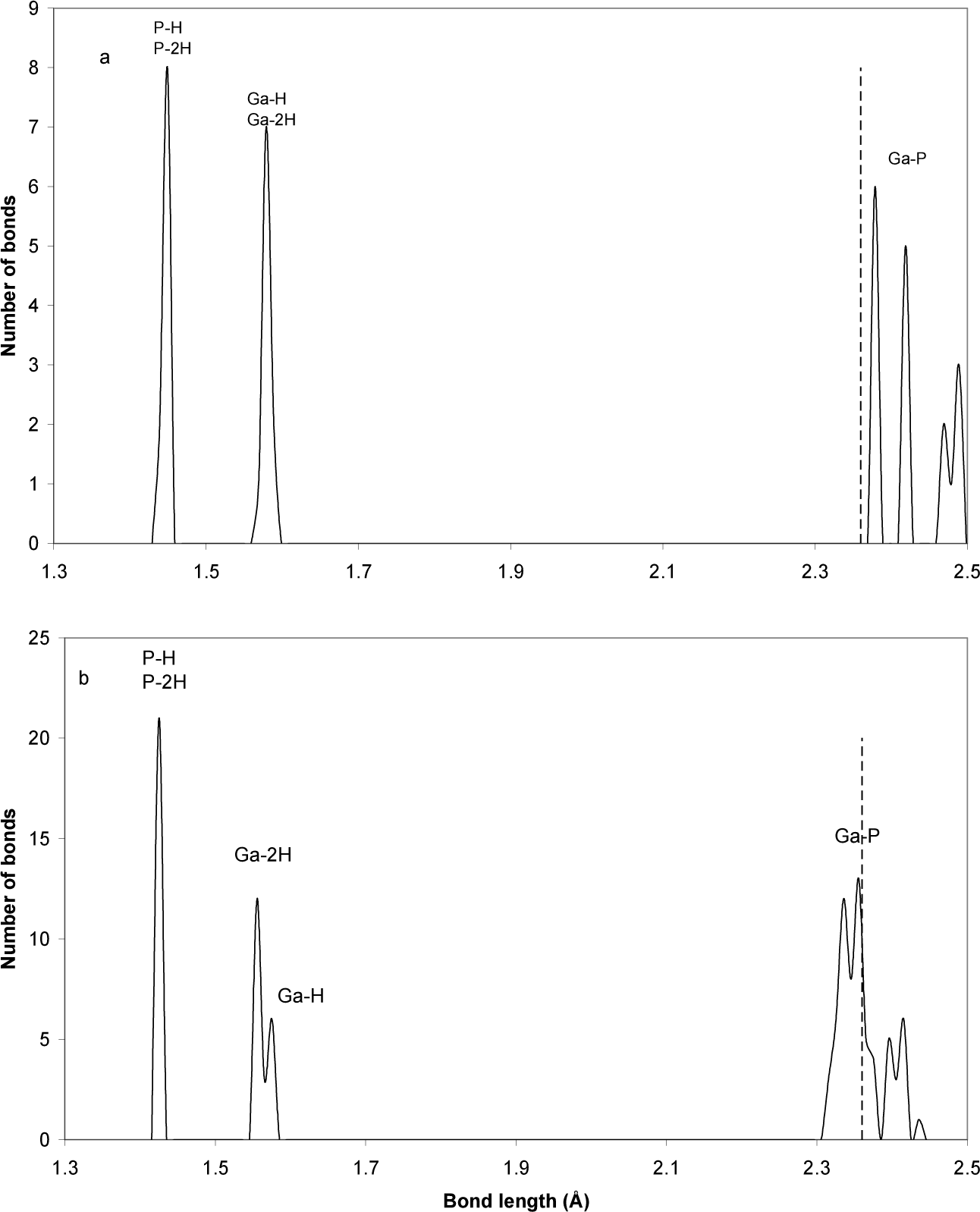
Distribution of bond lengths in (a) GaP-diamantane and (b) GaP-decamantane. The dashed line represents the experimental value of the GaP bulk bond length [3].
Fig. (6) shows the distribution of the values of GaP-diamantane and GaP-decamantane tetrahedral angles. The ideal bulk zincblende value of this angle at 109.47º is shown [14]. The distribution shows a narrow range around this angle (109.47º±13º), with the highest peak shifted one or two degrees from this value in decamantane. The GaP-diamantane angles are more distant from the ideal value than in the case of GaP-decamantane. Surface relaxation is responsible for this distribution, as it leads to the deviation of these angles from their ideal value. The same is true in Fig. (7) for the dihedral angle. The ideal bulk zincblende values of these angles are ±60 and ±180 [15]. Again, surface relaxation is responsible for the narrow deviation from these ideal values. The new peaks emerging for the GaP-decamantane are closer to the ±60 ideal value, which is not the case in GaP-diamantane.
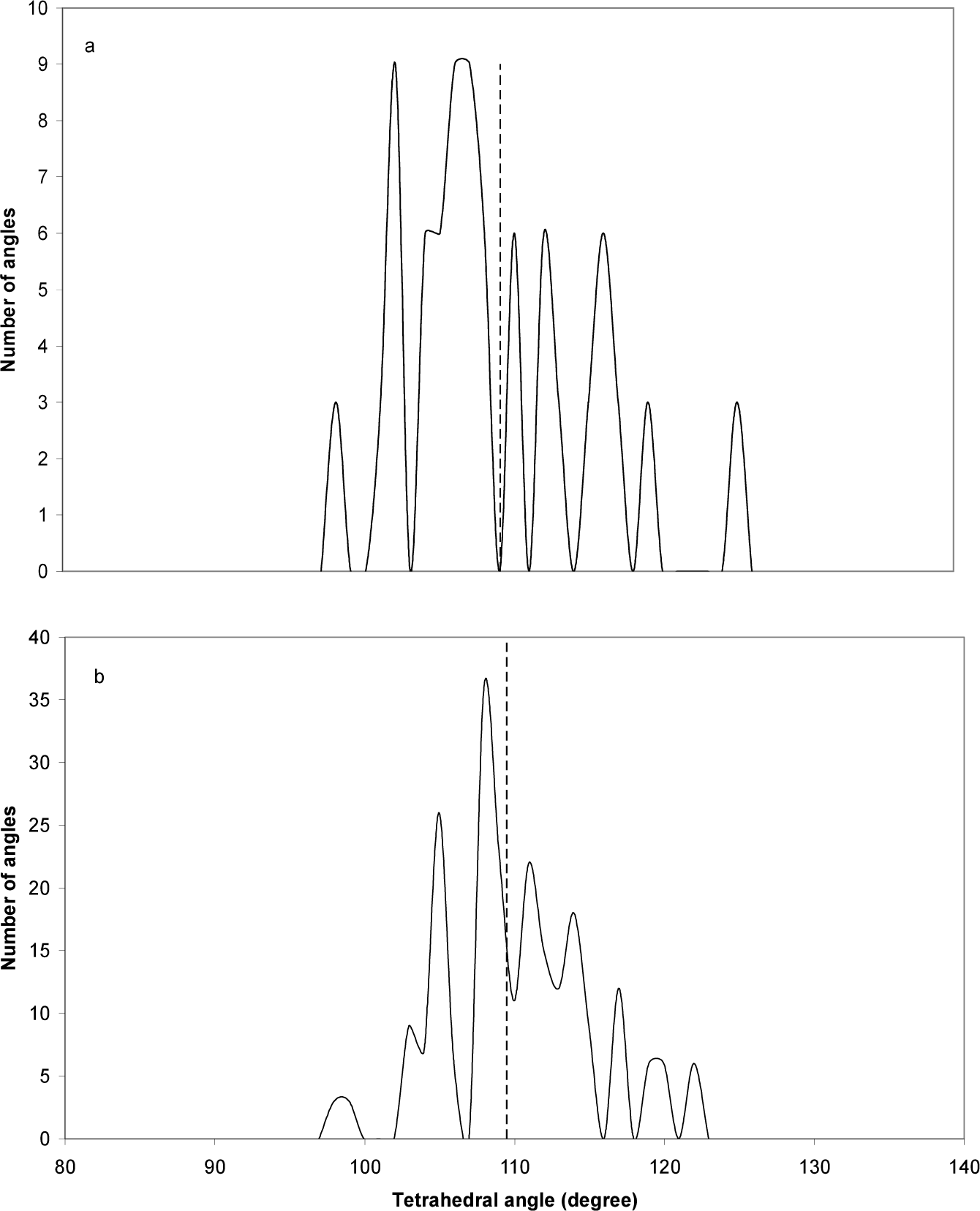
Distribution of the values of (a) GaP-diamantane and (b) GaP-decamantane tetrahedral angles. The dashed line represents the ideal bulk zincblende value of this angle, at 109.47º [14].
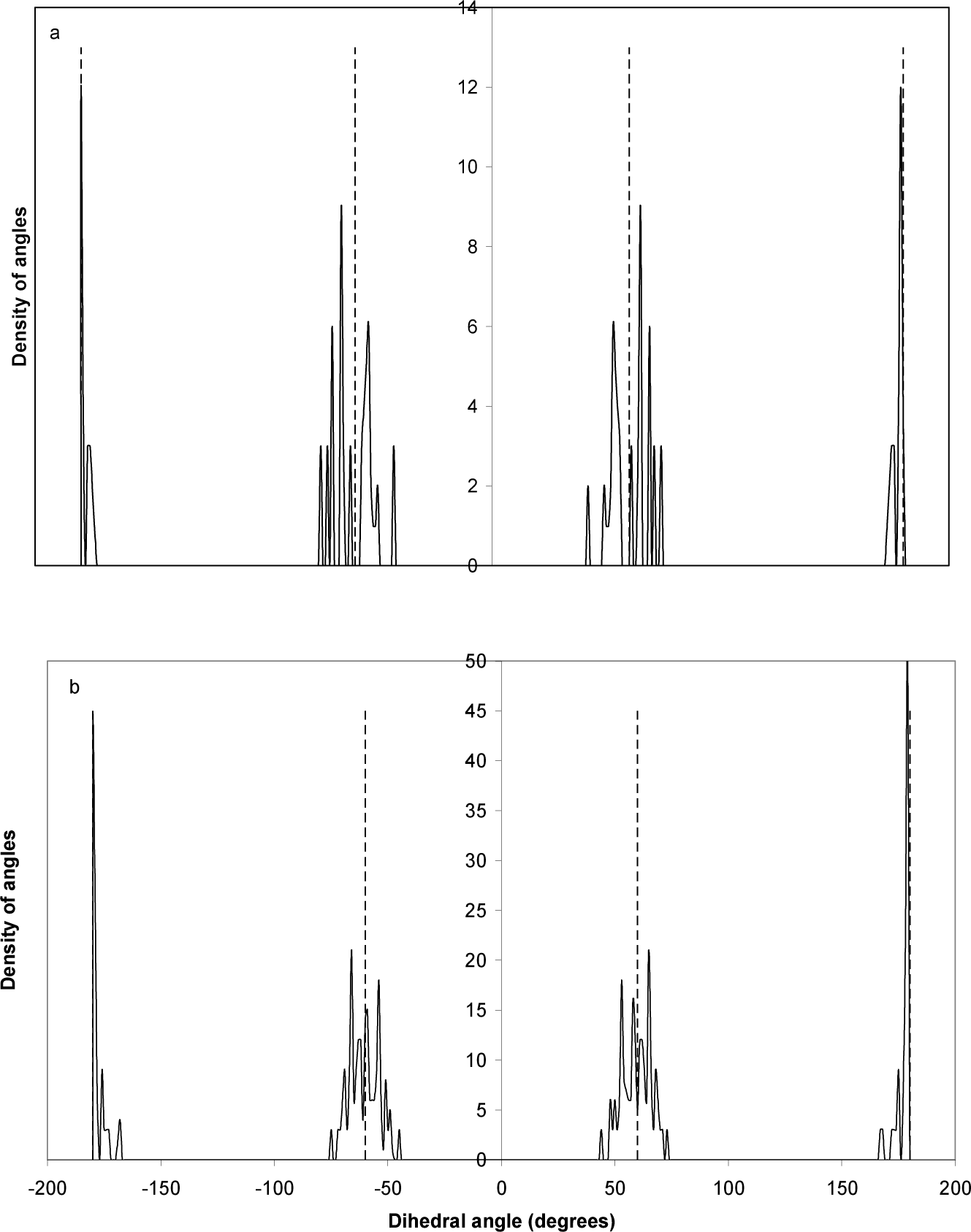
Distribution of (a) GaP-diamantane and (b) GaP-decamantane dihedral angles. The dashed lines represent the ideal values of zincblende structure, at −180º, −60º, 60º and 180º [15].
Figs. (8-11) are related to the vibrational spectroscopy of GaP diamondoids. In Fig. (8), the reduced masses of GaP-hexamantane molecular vibrations are shown. The dashed line represents the bulk experimental GaP longitudinal optical (LO) vibration frequency, at 376.9 cm−1 (11.3 THz) [3]. The highest reduced mass mode (HRMM) in Fig. (8), and the highest force constant mode (HFCM) [16, 17] in Fig. (9), at the longitudinal optical region, nearly coincides with the above-mentioned bulk experimental LO mode. This shows that diamondoids are a good representative of bulk GaP material. Although the IR peak associated with this mode (in Fig. (10)) is not high, this peak will increase in height as the size of these diamondoids increases, until it becomes the dominant peak at micrometre sizes. The corresponding Raman spectra share nearly the same properties as IR spectra, but with different selection rules [17].

Reduced masses of GaP-hexamantane molecular vibrations. The dashed line represents the experimental bulk GaP LO vibration frequency [3].

Force constant of GaP-hexamantane molecular vibrations. The dashed line represents the experimental bulk GaP LO vibration frequency [3].
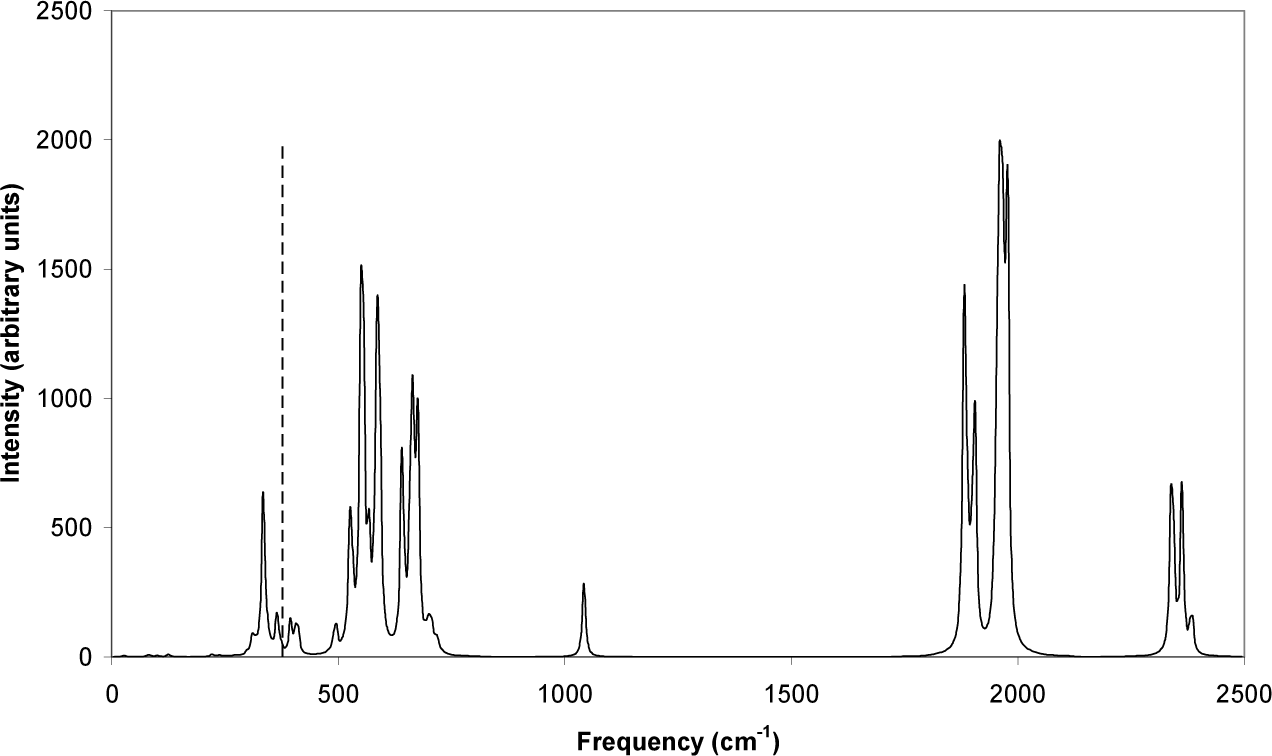
IR spectrum of GaP-hexamantane molecular vibrations. The dashed line represents the experimental bulk GaP LO vibration frequency [3].

Molecular size effects on various GaP diamondoid molecular vibrations, RBM, HRMM and asymmetric hydrogen surface vibration modes are shown. The dashed line represents the experimental bulk GaP LO vibration frequency [3].
The reduced mass of two particles of masses ma and mb is given by:

The reduced masses in Fig. (8) reach 30 atomic mass units (amu), which is nearly the reduced mass between two Ga atoms. This indicates that some vibrations might include one kind of atom but exclude the other. The vibrations after 471 cm−1 are all hydrogen-related vibrations, since they all have the approximate reduced mass of 1 amu. Although the reduced masses after 471 cm−1 are all nearly equal to 1 amu, the force constants in Fig. (9) keep on increasing, because the frequency of vibration is proportional to the square root of the force constant:

The type of vibration changes from the high reduced-mass vibrations of Ga-P in the region preceding 471 cm−1, to the symmetric and asymmetric Ga-2H and P-2H vibrations at around 1900 and 2400 cm−1 respectively, at the end of Figs. (8-10). The highest force constant for the Ga-P vibrations (2.1 mDyne/Å) is between that of germanium and silicon, as expected for GaP [18, 19] (germanium and silicon are adjacent, and share the same rows as gallium and phosphorus respectively, on the periodic table).
The GaP diamondoid size variation of four selected vibrations is shown in Fig. (11). These vibrations are radial breathing mode (RBM), longitudinal optical-highest reduced mass mode (LO-HRMM), and Ga-2H and P-2H asymmetric vibrations. The RBM can be identified by observing the atomic displacement of this vibration. Nearly all the atoms in this mode move in phase along the radius of the molecule [16]. The frequency of this vibration should converge to 0 cm−1 as we reach bulk sizes [20]. On the other hand, the LO-HRMM converges to 376.9 cm−1: as discussed above. The two modes meet at 187 cm−1 in the GaPH6 molecule. As a result, in moving from bulk to the nano-scale and molecular limit, the RBM is blue shifted while the LO-HRMM is red shifted. The two hydrogen-surface vibrational modes in Fig. (11) are nearly constant throughout the different sizes of GaP diamondoids and molecules. From this, we can suggest the following method to investigate the existence and size dependence of GaP diamondoids, First, we can be sure of the existence of these diamondoids from their constant distinguishable surface hydrogen vibrational modes; and second, we can determine the size of these molecules by analysing the Ga-P vibrational spectra in the region (0–471 cm−1), which can indicate frequency mode shifting from their corresponding bulk counterpart.
The number of atoms in the present molecules and nanocrystals does not exceed 100 atoms for the largest considered nanocrystal. This number of atoms is suitable for the kind of theory and basis set (PBE/6-31G(d)) used in the present work. To reach a higher number of atoms [21], one would need to use a less computationally expensive theory or basis. All the considered diamondoids are stable and have positive cohesive energies. The cohesive energy per atom of GaP-diamantane is 6.66 eV. This cohesive energy decreases as we proceed to heavier diamondoids, and it is reduced to 5.87 eV for GaP-decamantane. This value is less than the experimental value of bulk GaP (7.34 eV), which is the usual trend in DFT results [22].
4. Conclusions
GaP diamondoids are molecular/nano-particles with minimal surface effects, which make their electronic, structural and vibrational properties as close as possible to those of bulk zincblende structure. As a result, relatively small-sized GaP diamondoids can be used with good levels of accuracy to represent bulk or GaP nanocrystals. Diamondoids show minimal surface relaxation effects, which can be deduced from their bond lengths, tetrahedral angles and dihedral angles. Energy gap, bond length and vibrational modes are very close to those of their bulk counterparts. The IR vibrational spectra of GaP diamondoids are divided into two parts. The first part is mainly Ga-P vibrations; this part is located approximately within the range of 0–471 cm−1. This part is distinguished by a reduced mass that is larger than 1 amu. The second part – in the range 471–2400 cm−1 – is made up exclusively of hydrogen vibrational modes with a reduced mass very close to 1 amu. The first part encounters vibrational frequency shifts as the size of diamondoids change. RBM encounters blue shift as we move from bulk to the molecular or nano-scale; LO-HRMM encounters red shift as we move from bulk to the molecular or nano-scale. The two modes finally unite at 187 cm−1. The first part can be used to determine the size of the diamondoids. The second part, in the range 471–2400 cm−1: is nearly constant in its vibrational modes and can be used to identify GaP diamondoids.