
Abstract

Predicting and Treating Stress-Induced Vulnerability to Epilepsy and Depression
Becker C, Bouvier E, Ghestem A, Siyoucef S, Claverie D, Camus F, Bartolomei F, Benoliel JJ, Bernard. Ann Neurolog 2015;78:128–136.
Accumulation of stressful events can render individuals susceptible to develop epilepsy and comorbidities. Whether such vulnerability can be predicted and reversed is not known. Here we show that social defeat, although not producing depression by itself, produced in 50% of rats reduced threshold for status epilepticus (SE), accelerated epileptogenesis, and once epilepsy was induced, depression-like profile and cognitive deficits. Low serum brain-derived neurotrophic factor (BDNF) levels measured before SE identified this vulnerable population. Treatment with a BDNF analog before SE prevented the occurrence of comorbidities. Thus, vulnerability to comorbidities after epilepsy onset due to unresolved past stressful events may be predicted and reversed.
Commentary
Allostasis is defined as the process by which the body responds to stressors in order to regain homeostasis, which involves proper functioning of the hypothalamic-pituitary-adrenal (HPA) axis. Allostatic load refers to the pathophysiological consequences of dealing with excessive, prolonged, or unresolved stress and is also thought to involve abnormalities in the functioning of the HPA axis (for a review, see [1]). There has been a great deal of speculation regarding the role of allostatic load in disease, and numerous correlational studies have attempted to investigate the pathological implications of allostatic load. However, as the authors state in the highlighted manuscript, “causality remains to be determined.”
Through a series of studies, this group attempts to directly link allostatic load to pathophysiological outcomes. They previously demonstrated that social stress induces an allostatic load and vulnerability for depression in a subset of rats when faced with a subsequent, novel stressor (2). The authors suggest a “double-hit scenario” where the initial social defeat stressor (first hit) sensitizes a subset of animals, making them vulnerable to additional stressors (second hit). In the currently highlighted manuscript, Becker et al. extend these findings to investigate the pathophysiological implications of allostatic load acquired by exposure to a social defeat stress on epilepsy and associated comorbidities. This study, which is the basis of this Commentary, proposes that previous exposure to a stressor, which itself is insufficient to induce depression-like behavior, can alter epileptogenesis and predispose a subset of animals for associated comorbidities, including depression-like behaviors and cognitive impairments.
Stress has been implicated in epilepsy given that the majority of patients report that stress either triggers or worsens their seizures (3). Consistent with a role of stress in epilepsy, the currently highlighted study demonstrates that in a subset of rats (~50%) previous exposure to social stress results in a reduced threshold to induce status epilepticus (SE), a shorter latency to SE, and increased seizure frequency in chronically epileptic rats (see Figure 3 in the Becker et al. article). These results alone are not surprising, given the fact that stress has been demonstrated to increase seizure susceptibility in a variety of experimental epilepsy models (3) and has recently been demonstrated to accelerate epileptogenesis (4). Further, administration of exogenous stress hormones—including corticosterone and corticotropin-releasing hormone (CRH)—have been shown to exert proconvulsant actions in numerous experimental models of epilepsy (for a review, see [5]). However, these studies focus on how acute and prolonged stress or exposure to glucocorticoids can alter neuronal excitability and seizure susceptibility with little regard to how changes in the functioning of the HPA axis may influence epilepsy.
A large body of evidence suggests that early life stress can reprogram the HPA axis, altering HPA axis responsiveness, and increasing vulnerability for mood disorders as well as epilepsy (6). For example, maternal separation induces long-lasting effects on seizure susceptibility and increased propensity for epileptogenesis (for a review, see [7]). A recent study demonstrated that early life stress due to maternal separation exacerbated the seizure-induced elevations in corticosterone levels, reduced seizure threshold, and increased seizure severity (4). The vulnerability to epilepsy following maternal separation was shown to be due to reprogramming and hyperexcitability of the HPA axis (4). However, few studies have tackled the role of allostatic load in adult animals and the effect on HPA axis function and implications for disease.
To address the impact of allostatic load in adults, the authors of the reviewed study previously published that exposure to social defeat stress increases allostatic load in a subset of adult animals, making them vulnerable to subsequent chronic mild stress (CMS) and the development of depression-like behaviors (2). The vulnerable population was identified to have low serum levels of brain-derived neurotrophic factor (BDNF) and increased circulating levels of corticosterone. Treating animals with the BDNF analog, 7-8-dihydroxyflavone (7,8-DHF) or the antidepressant imipramine during the “second hit” prevented the elevations in corticosterone and the adverse behavioral consequences, including depression-like behavior and anhedonia. These data suggest that low levels of BDNF play a critical role in allostatic load in adult animals and vulnerability to depression. The manuscript that is the basis of this commentary builds on these initial studies to demonstrate that low serum BDNF levels following social defeat stress identifies a population of animals that are vulnerable to developing depression-like behaviors and cognitive impairments associated with epilepsy. The authors attempted to establish that following social defeat stress, prolonged reduction in serum BDNF levels is a diagnostic marker for this vulnerable population. Although it appears that this may be a useful diagnostic marker, it remains unclear whether this change is the real culprit in mediating the impact of stress on epilepsy and associated comorbidities.
Animals with low levels of BDNF also exhibit increased seizure activity, which could indirectly affect the associated comorbidities. The current study does not control for the effect of seizure frequency on comorbid behaviors and, therefore, seizure activity cannot be ruled out as a contributing factor. Further, rats with low BDNF levels also exhibit HPA axis hyperexcitability, which may contribute to the comorbidities associated with epilepsy. In fact, HPA axis hyperexcitability is only observed in animals that also exhibit depression-like behaviors and anhedonia. The authors attribute HPA axis hyperactivity to the phenotype of depression, which is justified, but it may also be part of the pathological process. Interestingly, treatment with the BDNF analog 7,8-DHF, normalized corticosterone levels as well as prevented the development of depression-like behaviors and cognitive impairments. It is tempting to speculate that BDNF may be playing a role in the regulation of the HPA axis and thereby influence seizure susceptibility and comorbidities associated with epilepsy. It would be interesting to assess whether blocking elevations in corticosterone alone could prevent increased seizure susceptibility and associated comorbidities as documented in the highlighted manuscript. For instance, blocking activation of the HPA axis with a corticosterone synthesis inhibitor has been demonstrated to prevent the effects of early life stress on seizure threshold and seizure severity (4), suggesting that pathological changes in HPA axis function underlie these pathophysiological consequences.
Interestingly, recent studies demonstrated that seizures themselves activate the HPA axis (4, 8), and seizure-induced elevations in corticosterone levels correlated with increased depression-like behaviors (9). It is possible that the “first hit” alters HPA axis reactivity, whereby in response to the “second hit,” there is hyperexcitability of the HPA axis that contributes to increased seizure susceptibility and associated comorbidities. In support of this hypothesis, only animals that exhibit depression-like behaviors associated with epilepsy also exhibit hyperactivation of the HPA axis following SE (see Figure 1 in Becker et al. article). Based on these studies, we propose a model whereby exposure to previous stressors induces an allostatic load, reprogramming the HPA axis in a subset of animals and leading to HPA axis hyperresponsiveness; this results in vulnerability when subjected to a “second hit,” at which time pathological consequences result, such as depression-like behaviors and increased seizure susceptibility (Figure 1).
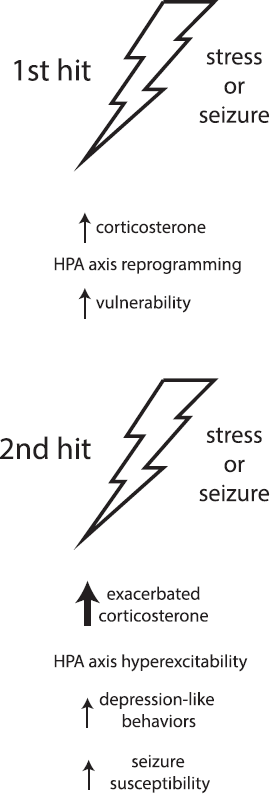
Despite the fact that this study does not definitively determine the pathophysiological mechanisms underlying the impact of stress on seizure susceptibility and associated co-morbidities, it does identify a potential biomarker of allostatic load and vulnerability, which was the intent of the current study and a significant contribution to the field. Future studies are required to determine the role of BDNF in the larger pathophysiological process leading to seizure susceptibility and associated comorbidities.