
Abstract

Mutations in PRRT2 Responsible for Paroxysmal Kinesigenic Dyskinesias Also Cause Benign Familial Infantile Convulsions [published online ahead of print March 8, 2012].
Ono S, Yoshiura KI, Kinoshita A, Kikuchi T, Nakane Y, Kato N, Sadamatsu M, Konishi T, Nagamitsu S, Matsuura M, Yasuda A, Komine M, Kanai K, Inoue T, Osamura T, Saito K, Hirose S, Koide H, Tomita H, Ozawa H, Niikawa N, Kurotaki N. J Hum Genet 2012. doi:10.1038/jhg.2012.23.
Paroxysmal kinesigenic dyskinesia (PKD (MIM128000)) is a neurological disorder characterized by recurrent attacks of involuntary movements. Benign familial infantile convulsion (BFIC) is also one of a neurological disorder characterized by clusters of epileptic seizures. The BFIC1 (MIM601764), BFIC2 (MIM605751) and BFIC4 (MIM612627) loci have been mapped to chromosome 19q, 16p and 1p, respectively, while BFIC3 (MIM607745) is caused by mutations in SCN2A on chromosome 2q24. Furthermore, patients with BFIC have been observed in a family concurrently with PKD. Both PKD and BFIC2 are heritable paroxysmal disorders and map to the same region on chromosome 16. Recently, the causative gene of PKD, the protein-rich transmembrane protein 2 (PRRT2), has been detected using whole-exome sequencing. We performed mutation analysis of PRRT2 by direct sequencing in 81 members of 17 families containing 15 PKD families and two BFIC families. Direct sequencing revealed that two mutations, c.649dupC and c.748C4>T, were detected in all members of the PKD and BFIC families. Our results suggest that BFIC2 is caused by a truncated mutation that also causes PKD. Thus, PKD and BFIC2 are genetically identical and may cause convulsions and involuntary movements via a similar mechanism.
Commentary
Three inherited paroxysmal neurologic disorders have all been mapped to the pericentromeric region of human chromosome 16. Paroxysmal kinesigenic dyskinesia, also known as episodic kinesigenic dyskinesia (EKD), is an adolescent-onset disorder characterized by brief, recurrent episodes of involuntary movement triggered by sudden voluntary movement (MIM128200). EKD1 was mapped to 16p11.2-q12.1 by linkage analysis in eight Japanese families (1), some of which were used for the current study by Ono and colleagues. Benign familial infantile seizures (BFIS) is characterized by infant-onset, afebrile seizures that remit by 2 years of age (MIM605751). BFIS2 was mapped to 16p12-q12 by linkage analysis in seven families (2). Co-occurrence of EKD and infantile seizures is referred to as infantile convulsions and choreoathetosis (ICCA) syndrome. Within the same family, individuals may have infantile seizures, EKD, or both (MIM 602066). Linkage analysis in four French families mapped ICCA to 16p12-q12 (3). Based on the paroxysmal nature of these disorders, the co-occurrence of EKD and infantile seizures in some families, and the overlapping map positions, it has been hypothesized that these disorders are allelic.
In late 2011, PRRT2, encoding proline-rich transmembrane protein 2, was identified as the gene responsible for EKD1 by whole exome sequencing in eight Han Chinese families (4). Concurrently, Wang and colleagues reported truncating PRRT2 mutations in five EKD1 families, including two large pedigrees with co-occurring EKD and infantile seizures (5). In early 2012, Heron and colleagues reported PRRT2 mutations in a number of ICCA families, as well as families with BFIS only (6). They identified PRRT2 mutations in 14 of 17 BFIS families (82%) and in 5 of 6 ICCA families (83%). Together, these studies provided evidence that EKD1, ICCA, and BFIS2 are, in fact, allelic.
PRRT2 is a four-exon gene that encodes a 340 amino-acid protein with two transmembrane domains. PRRT2 is widely expressed in the developing and adult central nervous system, with high levels of expression in the cerebral cortex and basal ganglia (4, 6). It has been shown to interact with SNAP25, a presynaptic protein involved in neurotransmitter release (7, 8). Most PRRT2 mutations reported to date are nonsense mutations that result in premature stop codons. Although the exact function of PRRT2 is unknown, determining the mechanistic basis of PRRT2 mutations may provide new insights into the pathogenesis of infantile epilepsy.
In the present study, Ono and colleagues examined 17 Japanese families, including 7 families with EKD only, 2 families with BFIS only, and 8 ICCA families. Overall, this included 68 PKD patients and 13 patients with infantile seizures. To screen for PRRT2 mutations in the patient samples, they performed PCR amplification and Sanger sequencing of PRRT2 exons. In 16 of 17 families (94%), they observed a common mutation, c.649dupC which results in the frameshift mutation p.R217PfsX8. Observation of the same mutation in 16 of 17 Japanese families suggested the possibility that c.649dupC could be a founder mutation. However, by haplotype analysis there were six different haplotypes in the PRRT2 region in these families, making it less likely to be a founder mutation. Additionally, the c.649dupC mutation has been reported in several other studies in different populations, including Han Chinese, European, Australian, Ashkenazi Jewish, and Sephardic Jewish populations (4–6, 9). Together, these observations suggest that this is a mutation hot spot. The c.649dupC mutation occurs in the context of a homopolymer repeat of nine cytosines, which may be susceptible to slipped-strand mispairing during DNA replication (Figure 1). Based on the available studies, the c.649dupC mutation in PRRT2 accounts for 50 to 75 percent of PKD, 50 to 80 percent of ICCA and 80 to 86 percent of BFIS (4–6, 8–10).
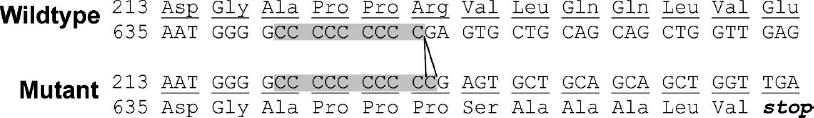
A hot-spot mutation in the PRRT2 gene. Insertion of a cytosine at cDNA position 649_650 results in the frameshift mutation R217PfsX8. This mutation occurs in the context of a homopolymer repeat in a GC-rich region, which may be susceptible to slipped-strand mispairing during DNA replication.
The most novel and significant finding in this study was the observation of c.649dupC as a de novo mutation in one family. In a three-generation family, the c.649dupC mutation arose in the second generation in an individual with PKD who subsequently transmitted the mutation to a child diagnosed with benign infantile seizures. The observed de novo mutation suggests that PRRT2 mutations may contribute to sporadic benign infantile seizures, as well as EKD.
The significant contribution of PRRT2 mutations to EKD, BFIS, and ICCA along with the occurrence of a mutation hot spot in PRRT2 suggests that screening of patients for the c.649dupC prior to additional testing may be an efficient and cost-effective strategy for molecular diagnosis. Further, the observation of a de novo mutation of PRRT2 suggests that screening of PRRT2 should be considered for sporadic benign infantile seizures.