
Abstract
The diagnosis of renal amyloidosis is sometimes elusive. In this manuscript, we present and discuss two cases, presenting with proteinuria, subsequently diagnosed with rare variants of renal amyloidosis: AH amyloidosis in patient with lymphoplasmacytic lymphoma, and AA amyloidosis in patient with hyperimmunoglobulinemia D syndrome.
Keywords
Case 1
Case description
A 41-year-old woman was referred to our clinic in January 2010 for a consultation. Her main complaints were general weakness; physical examination did not reveal any abnormalities. Previous medical history included weakness, night sweats and microscopic hematuria since 2005. In 2009, moderate proteinuria (1.27 g/day), mild anemia (10.4 g/dL) and splenomegaly were found. Workup for lupus in local nephrology unit was negative, and bone marrow aspirate smear showed 6.5% plasma cells. Kidney biopsy revealed by light microscopy 2 ischemic glomeruli out of 22, while in the rest of the glomeruli there were no proliferative changes. Mesangium widening and glomerular basement membranes and arteriolar walls irregular thickening with homogenous slightly PAS-positive, strongly Congo red–positive material with typical birefringence under polarization were revealed. Immunohistochemistry of formalin-fixed paraffin-embedded tissue with immunoperoxidase staining for amyloid A protein was negative, which led to a diagnosis of amyloidosis of unknown origin. We were requested to clarify the diagnosis and recommend adequate treatments.
Workup in our unit showed proteinuria 0.34 g/day, urine red blood cells 8-10-50-60 high-power field, Hb 12.1 g/dL with normal total blood cell count, erythrocyte sedimentation rate (ESR) 13 mm/hour, and blood chemistry otherwise normal. Abdomen and kidney ultrasound revealed nothing but moderate splenomegaly of 157 × 57 × 110 mm, 77 cm2.
Given hematuria as a first symptom and moderate proteinuria, the most obvious preliminary diagnosis would be IgA nephropathy. But this was not consistent with anemia – as her kidney function was normal, and no iron deficiency was found – or with kidney pathology findings. Splenomegaly and slight elevation in bone marrow of plasma cell count also could not be explained in the setting of IgA nephropathy.
Additional workup included skeletal X-ray and chest and abdomen computed tomography (CT), which were both unremarkable, and serum and urine electrophoresis, which revealed G
The second kidney biopsy (7 glomeruli were found by light microscopy to be without proliferative changes) showed deposition of eosinophilic, amorphous, weakly PAS-positive, Congo red-positive material in mesangium (Fig. 1), capillary walls and arteriolar walls. Sections stained with Congo red were examined in polarized light and demonstrated typical apple-green birefringence. Immunofluorescent study of unfixed frozen tissue for IgA, IgM, IgG, C3, C1q and
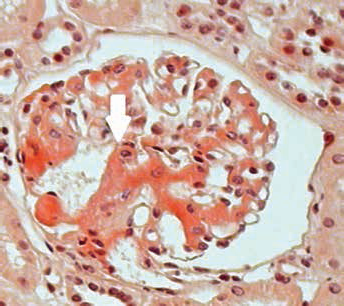
Light microscopy showing renal amyloidosis, with Congo red-positive homogenous material in mesangium (staining: Congo red; magnification ×200).
As we failed to prove AL amyloidosis but found instead AH amyloidosis, we performed a repeated search of the source of monoclonal heavy chains. Repeated serum and urine electrophoresis found heavy chain γ paraproteinemia and traces of Bence Jones
Based on clinical presentation with weakness, night sweats, anemia and splenomegaly and the workup showing lymphoplasmacytic infiltration of bone marrow, presence of paraprotein heavy chain γ, secondary immunodeficiency and kidney amyloidosis with positive immunofluorescence for IgG, the patient was diagnosed with gamma heavy chain disease (γ-HCD; also known as Franklin's disease) with AH amyloidosis. She was successfully treated in the Hematology Research Institute with a combination of rituximab, bortezomib and dexamethasone, and her treatment was subsequently consolidated with high-dose chemotherapy and autologous stem cell transplantation (ASCT).
Background
AH amyloidosis, a part of the continuum of immunoglobulin-related amyloidosis, is an exceptionally rare condition, first described in 1990. Similar to AL, AH amyloidosis is associated with MGUS and plasma cell/B-cell lymphoproliferative disorders, with only a few cases derived either from truncated γ or μ heavy chains reported so far. The most recent paper describes 5 patients with AH and 11 patients with AL/AH amyloidosis (1–4).
Heavy chain diseases are B-cell proliferative disorders characterized by production of abnormal, structurally incomplete, immunoglobulin heavy chains without corresponding light chains; abnormal heavy chains are the result of gene mutations. γ-HCD is a type of heavy chain disease, a lymphoplasmacytoid lymphoma characterized by production of incomplete monoclonal γ heavy chains without associated light chains. Clinical presentation most commonly resembles that of patients with systemic lymphoproliferative/autoimmune diseases. Prevalence is unknown, and age at diagnosis of γ-HCD ranges from 42 to 87 years, with a median of 68 years. There have only been about 120 cases reported in the literature worldwide, but this condition is thought to be underdiagnosed. We found only 1 description of AH amyloidosis associated with γ-HCD (5, 6).
Case 2
Case description
A 33-year-old woman was referred to our clinic in July 2007 with general weakness, fever and headache. Physical examination at admission was unremarkable, except macular face rush and mild cervical lymphadenopathy.
She was known to experience recurrent episodes of fever since early childhood, along with cervical lymphadenopathy, arthralgia, leukocytosis and elevated C-reactive protein (CRP), which was primarily considered as rheumatism and treated with bicillinum. At age 15, after 1 of multiple fever episodes, mild proteinuria, microscopic hematuria, elevated white blood cell count and ESR were found. She was tested for lupus (negative), rheumatoid arthritis (negative) and toxoplasmosis (positive), and treated with co-trimoxazole. At age 22, after pregnancy, which was terminated at 8 weeks due to a typical episode of fever and arthralgia, recognized as flu, she developed facial edema, mild arterial hypertension, anemia and leukocytosis, and her proteinuria increased up to 4.2 g/L, and CRP up to 133 mg/L Serology for lupus, rheumatoid arthritis and vasculitis was negative.
She was diagnosed with chronic glomerulonephritis (not otherwise specified) by kidney biopsy, performed in the local general hospital, received nephroprotective treatment and continued to experience recurrent fever and arthralgia. Proteinuria ranged from 5.0 to 0.5 g/L with total protein 5.4 g/dL, and her blood pressure did not exceed 140/90 mm Hg.
Ten years later, a second biopsy was performed in the regional hospital, and showed by light microscopy eosinophilic Congo red–positive material, infiltrating glomeruli and arterial walls, which led the local nephrologists, taking into consideration her arthralgia, to the diagnosis of seronegative arthritis, complicated by amyloidosis. She was advised to take leflunomide, and referred to our clinic for a second opinion.
Workup showed moderate proteinuria of 0.7–1.8 g/day, serum creatinine 180 μmol/L, urea 9,5 mmol/L and total protein 5.2 g/L. Other blood chemistry tests and total blood count were normal. CRP exceeded the upper normal level by 6 times, RF was negative and C-3 complement within the normal range. Serology tests for toxoplasmosis were negative. Serum and urine immunochemistry showed elevated IgA and β2-microglobulin, decreased IgG and nonselective glomerular proteinuria. Chest, extremities, spine and pelvis X-ray, abdomen and kidney ultrasound, chest and abdomen CT, sacroiliac magnetic resonance imaging (MRI) and colonoscopy did not show any significant changes.
Moderate proteinuria with microscopic hematuria since age of 15, already impaired kidney function without developing of nephrotic syndrome, elevated serum IgA and CRP, and findings of the first and second kidney biopsies could be explained again by IgA nephropathy with secondary focal segmental glomerulosclerosis, falsely interpreted as amyloidosis, in a patient with chronic tonsillitis. On the other hand, recurrent fever, arthralgia, cervical lymphadenopathy and macular face rush did not allow us to exclude lupus, despite negative previous workup.
To differentiate one from the other, paraffin blocks from both kidney biopsies were reprocessed. The total number of glomeruli by light microscopy was up to 30, with 6 of them totally sclerosed. The rest looked enlarged, with homogenous Congo red–positive staining with typical birefringence, and Jones silver–negative material localized in mesangium, subepithelial and subendothelial space (Fig. 2) and small artery walls. Immunofluorescence of formalin-fixed paraffin-embedded sections with FITC-conjugated anti IgA, IgG, IgM, C1q, C3, fibrinogen, λ and
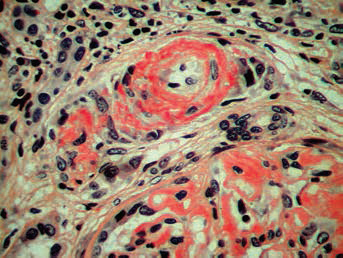
Light microscopy showing renal amyloidosis, with Congo red-positive homogenous material in mesangial and subendothelial space and arteriolar walls (staining: Congo red; magnification ×100).
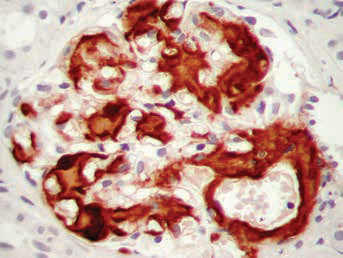
Light microscopy showing renal amyloidosis, with prominent expression of amyloid A component in mesangial and subendothelial space and arteriolar walls (staining: immunoperoxidase, magnification ×100).
Given the diagnosis of AA amyloidosis, and the recurrent fever, arthralgia, lymphadenopathy, leukocytosis and elevated CRP since early childhood, we suspected autoinflammatory syndrome. The patient underwent genetic testing for MEFV mutations, but no mutations in the 10 exon were found. Subsequent testing for MVK gene mutations found 2 mutations: heterozygous p.Val377lnapoc.2015.14613le mutation in exon 11 and heterozygous p. Trp188Gly mutation in 6 exon, which was suggestive for diagnosis of hyper-IgD syndrome. Her IgD level was found to be normal, but IgA was again elevated one-half the upper normal level. The patient was diagnosed with hyper-IgD syndrome, received only supportive treatment, but within 3 years slowly progressed to end-stage renal disease and started continuous ambulatory peritoneal dialysis treatment in 2013.
Background
In AA amyloidosis amyloid fibrils are composed of fragments of serum amyloid A (SAA) protein, a major acute-phase reactant protein. AA amyloidosis occurs in the course of a chronic inflammatory disease of either infectious or noninfectious etiology, hereditary periodic fevers, and with certain neoplasms such as Hodgkin disease and renal cell carcinoma. In developing countries, the most common instigator of AA amyloidosis is chronic infection; in industrialized societies, rheumatic diseases, such as rheumatoid arthritis, are the usual stimuli (7).
Hereditary periodic fever syndromes/autoinflammatory syndromes are rare and distinct heritable disorders characterized by short and recurrent attacks of fever and severe localized inflammation that occur periodically or irregularly and that are not explained by usual childhood infections. These attacks undergo spontaneous remission without antibiotic, antiinflammatory or immunosuppressive treatment. Between attacks, patients feel well and regain their normal daily functions until the next episode occurs. The episodes are usually associated with elevated serum levels of acute-phase reactants (e.g., CRP, fibrinogen, SAA), elevated ESR and leukocytosis. Six periodic fever diseases have been well characterized over the last few years, and considerable recent progress has been made in identifying causative genes and developing treatment options These 6 disorders are known as: (i) familial Mediterranean fever (FMF); (ii) hyperimmunoglobulinemia D with periodic fever syndrome (HIDS); (iii) tumor necrosis factor (TNF) receptor-associated periodic syndrome (TRAPS); (iv) Muckle-Wells syndrome (MWS); (v) familial cold autoinflammatory syndrome (FCAS) and (vi) chronic infantile neurologic cutaneous articular syndrome (CINCA), also known as neonatal-onset multisystem inflammatory disease (NOMID) (8–10).
HIDS, also known as mevalonate kinase deficiency (MKD), was firstly described in 1984 in a Dutch family. The patients had a long history of recurrent attacks of fever of unknown cause, as well as high serum IgD levels and numerous plasma cells with cytoplasmic IgD in the bone marrow. Clinical symptoms during the attacks included lymphadenopathy, abdominal pain, diarrhea, headache, hepatomegaly and/or splenomegaly, arthralgia and/or arthritis and skin lesions. Later, the disease was identified in patients from other European countries such as England, Germany, Italy, Turkey and the Czech Republic, as well as the United States and Japan. The disease has an early age of onset. Most patients have attacks before the end of their first year of life (median 0.5 years). The attacks persist throughout life, although patients have a reduction in intensity and frequency of attacks after adolescence. (11–15).
Diagnostic criteria for HIDS are as follows: constant high IgD level (>100 U/mL) measured on 2 occasions at least 1 month apart; during attacks, elevated ESR and leukocytosis, abrupt onset of fever (temperature at least 38.5°C), recurrent attacks, elevated IgA level, cervical lymphadenopathy, abdominal distress (vomiting, diarrhea, pain), skin manifestations (erythematous macules and papules), arthralgia and/or arthritis, splenomegaly. The most typical finding is the consistently elevated serum IgD level, although patients can have normal IgD levels. A normal IgD level does not exclude the diagnosis. Approximately 82% also have elevated serum IgA levels (16, 17).
Renal AA amyloidosis is a virtually universal complication of FMF in some populations if patients are not compliant with colchicine prophylaxis. Other hereditary fever syndromes, such as TRAPS, CINCA, MWS and HIDS, may be complicated by AA amyloidosis, but not as often. We found only a few descriptions of AA amyloidosis in HIDS (18–22).
Discussion
Difficulties usually experienced in diagnostics of amyloidosis are determined by its nature, as most often amyloidosis is in fact a complication of various diseases: lymphoplasmacytic, chronic inflammatory, infectious, malignant etc. Diagnosis usually comes late, as the main disease either is overt and masking symptoms of amyloidosis or on the contrary, is subtle and underrecognized, while amyloidosis shows its symptoms only when target organs are seriously damaged. In both of the cases presented here, absence of nephrotic syndrome, basicly typical for renal amyloidosis, made the diagnosis even more complicated, and only repeated/reprocessed kidney biopsy, confirming the diagnosis of amyloidosis as such and determining its type, targeted the search to specific diseases as being the cause of renal amyloidosis.
Treatment of amyloidosis is aimed to eliminate precursors of amyloid, depending on the underlying disease. For γ-HCD, chemotherapy, mostly using agents efficacious in lymphoma and multiple myeloma (e.g., cyclophosphamide, prednisone, vincristine, chlorambucil, doxorubicin), has been disappointing. However, partial and short-lived responses have been reported using fludarabine and/or rituximab. There are also reported uses of rituximab in nephrotic syndrome in patients with AH amyloidosis (23–26). However, in our case 1, treatment with rituximab, bortezomib and dexamethasone with subsequent consolidation by high-dose chemotherapy and ASCT was effective. Treatment of HIDS so far is largely supportive because various standard antiinflammatory drugs (including colchicine and steroids) fail to suppress the attacks. A trial with thalidomide showed only a nonsignificant decrease of acute phase protein synthesis but without an effect on the attack rate. In another study, simvastatin usage resulted in a decrease in the urinary mevalonic acid concentration in all patients and decreased the number of febrile days in 5 of 6 patients (27, 28). In case 2, proper diagnosis allowed the avoidance of useless treatment with leflunomide.
Outcomes depend on the efficacy of eliminating amyloid precursors and also on the severity of organ damage at the time of diagnosis. Thus, early diagnosis is crucial, for this clinicians have to consider amyloidosis in all cases of adult nephrotic syndrome, and as both presented cases depict – also in cases of moderate proteinuria. Kidney biopsy with Congo red staining and immunofluorescence/immunohistochemistry is the gold standard of diagnostics.
Footnotes
Financial support: None.
Conflict of interest: None.