
Abstract
Amyloidosis is a heterogeneous group of hereditary and acquired diseases in which normally soluble plasma proteins are deposited in the extracellular and/or intracellular space in abnormal, insoluble, fibrillar form. Renal damage is one of the most common features of systemic amyloidosis, and the presentation is most commonly due to the consequences of renal involvement, with proteinuria and progressive renal decline. Progression to end-stage renal failure is common. Early diagnosis of systemic amyloidosis is difficult. Renal amyloidosis typically presents with nephrotic syndrome and/or renal failure.
Treatment of AL amyloidosis aims to reduce production of the monoclonal immunoglobulin precursorvia chemotherapy. Current options for treatment include melphalan+dexamethasone or cyclophosphamide-bortezomib-dexamethasone regimens, or in selected patients, high-dose melphalan with autologous stem cell transplantation. The focus of current research is on pharmacological therapy to solubilize amyloid fibrils and increase tissue catabolism of amyloid deposits.
Introduction
Amyloidosis is not a single disease – it represents a heterogeneous group of diseases or complications in which normally soluble plasma proteins are deposited in the extracellular and/ or intracellular space in abnormal, insoluble, fibrillar form. Accumulation of these fibrils leads to progressive impairment of tissue and organ structure and function. Ninety percent of amyloid is constituted by major fibrillar amyloid proteins, and another 10% includes glycosaminoglycans, serum amyloid P-component and apolipoprotein E. Diagnosis is based on presence of amyloid in tissues, proven by pathology. Light microscopy with hematoxylin-and-eosin staining shows amorphous eosinophilic masses, and the gold standard of diagnostics is Congo red staining of amyloid in salmon-pink, with apple-green birefringence under polarized light. This birefringence is defined by the specific β-pleated sheet architecture of amyloid. The fibrillar nature of amyloid is visible by electron microscopy.
Types of amyloidosis
The type of amyloid is defined by the nature of the mis-folded proteins making up the amyloid fibrils. The current classification system is based on the chemical properties of the amyloid, using abbreviations with initial capital A for amyloid as such, followed by capitals that are abbreviations for fibril proteins: for example, light-chain amyloidosis is abbreviated AL, heavy-chain amyloidosis as AH, serum A protein amyloidosis as AA etc. Currently more than 30 proteins that form amyloid fibrils are recognized (see Tab. I).
Major types of amyloidosis
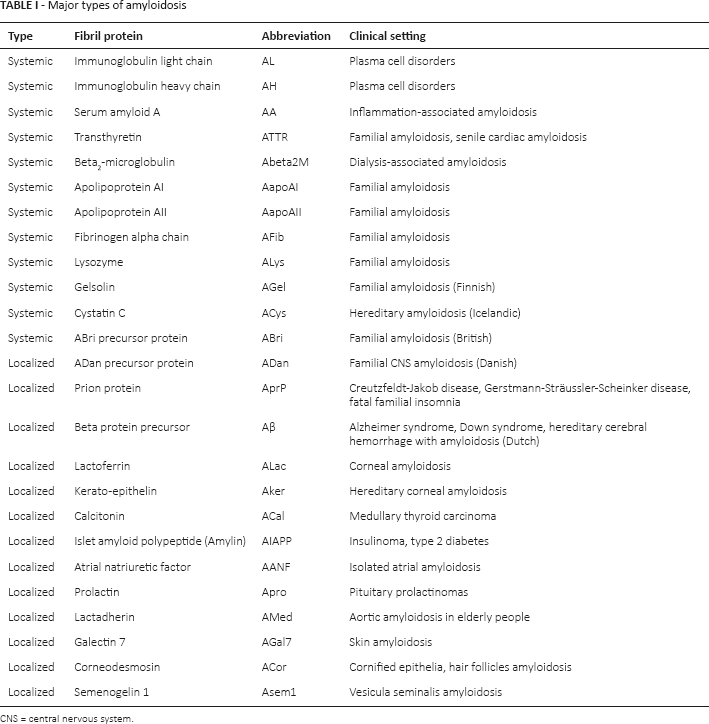
CNS = central nervous system.
Renal damage is one of the most common features of systemic amyloidosis (AL, AH, AA, ATTR). Systemic AL amyloidosis, beyond the kidneys, typically involves also heart, peripheral nerves, gastrointestinal tract, respiratory tract and nearly any other organ. Kidneys, liver and spleen involvement is characteristic for AA amyloidosis. There are a few cases of hereditary amyloidosis with primarily kidney involvement (AFib, AapoAl, AapoAll and ALys) described so far (1–6).
AL/AH amyloidosis (or immunoglobulin-related amyloidosis) resulting from fibrillar deposition of monoclonal proteins, mostly light chains, is a part of a group of monoclonal plasma cell disorders, such as multiple myeloma and related diseases. AL amyloidosis may occasionally develop in patients with multiple myeloma or lymphoplasmacytic lymphoma, but mostly is associated with low-grade plasma-cell clone and does not meet diagnostic criteria for any of these condition (so-called “primary AL amyloidisis”). This term, as well as the term monoclonal gammopathy of undetermined significance (MGUS) used to be applied, until recently the term monoclonal gammopathy of renal significance (MGRS) was intoduced for AL amyloidisis and other types of renal damage due to monoclonal protein deposition in patients without overt plasma cell malingnancies (7–9).
AA amyloidosis is associated with various chronic inflammatory disorders, chronic local or systemic microbial infections, and occasionally with neoplasms. The precursor protein is serum amyloid A protein, which is an acute phase reactant produced mainly in the liver in response to multiple cytokines. The spectrum of these diseases is shown in Table II. Clinical presentation is most commonly due to the consequences of renal involvement, with proteinuria and progressive renal decline. Progression to end-stage renal failure is common (10, 11).
Main causes of AA amyloidosis
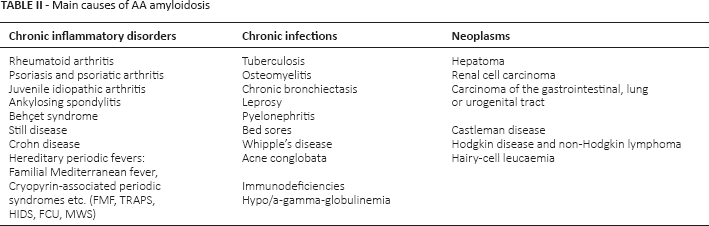
AL and AA amyloidosis are the most common types of amyloidosis worldwide. Frequency of AL amyloidosis is about 1–5 cases/100,000 inhabitants, but the prevalence of AA amyloidosis is difficult to estimate because of the heterogenicity of the causative diseases.
Diagnosis
Early diagnosis of systemic amyloidosis is difficult, as symptoms of kidney and other, often multiorgan involvement, basicly present in the late stages with prominent amyloid deposition. Renal amyloidosis typically presents with nephrotic syndrome and/or renal failure. For AL amyloidosis, the clue to diagnosis may be the presence of involvement of the heart and the peripheral vasculature with postural hypotension, weakness, palpitations, dyspnea, congestive heart failure and arrhythmias along with nephrotic syndrome. Patients with involvement of the peripheral nervous system often present with dysesthesia, decreased sensation and decreased strength. Gastrointestinal symptoms include nausea, vomiting, gastrointestinal hemorrhage, obstruction, diarrhea and constipation, or alternating constipation and diarrhea. An important indication is arterial hypotension, in particular in previously hypertensive patients. For AA amyloidosis, the presence of nephrotic syndrome in patients with chronic inflammatory disorders or chronic infections demands the consideration of amyloidosis. In any case, a kidney biopsy with Congo red staining is mandatory to confirm diagnosis. Defining of the type of amyloid, using immunofluorescence/immunohistochemistry with a panel of standard antibodies, including anti-λ and anti-
Treatment
Treatment of AL amyloidosis aims to reduce the production of the monoclonal immunoglobulin precursor via chemotherapy. Current treatment, targeting causal B-cell clones, based on extrapolation of treatments for overt malignancy, include melphalan + dexamethasone or cyclophosphamide-bortezomib-dexamethasone regimens, or in selected patients, high-dose melphalan with autologous stem cell transplantation.
In AL amyloidosis, survival depends on hematological response to therapy, on the extension and severity of organ involvement and on the presence or not of amyloid heart disease. The worst prognosis is associated with clinical symptoms of cardiac involvement, with a median survival rate of 6 months. Prognosis is not influenced by the underlying plasma cell proliferation. However, identification of a neoplastic plasma cell population adversely affects survival, and a bone marrow plasma cell infiltration above 10% has been associated with poorer outcome. Patients with involvement limited to the peripheral nerves have the longest survival. Other favorable prognostic factors include normal renal function. For renal amyloidosis, a meaningful clinical response is defined as a 50% decrease (at least 0.5 g/day) of 24-hour urine protein in the absence of a reduction in estimated glomerular filtration rate (eGFR) ≥25% or an increase in serum creatinine ≥0.5 mg/dL. AL amyloidosis is a serious disease and causes death when treatment is delayed, whereas new therapeutic strategies induce hematological remission in most patients, with a median survival of more than 5 years. In the absence of chemotherapy, systemic AL amyloidosis is always progressive. Early diagnosis is therefore a critical step in the care of these patients (12–15).
Treatment of AA amyloidosis is traditionally aimed at the underlying inflammatory condition to reduce the production of the precursor amyloid – serum A protein. Monitoring of the serum amyloid A protein is vital to assess whether there is adequate suppression of the underlying disease. The level of serum amyloid A protein is a powerful predictor of both patient survival and renal outcome. In patients with adequate suppression of the serum amyloid A protein, amyloid deposits can be seen to regress, and renal function can be stabilized and even improved. The prognosis for AA amyloidosis regardless of the prognosis of the primary disease is associated with the degree of renal damage at the time of diagnosis, with poor prognosis associated with a serum creatinine level greater than 2 mg/dL or a serum albumin level of less than 2.5 g/dL. Mean patients survival is 2–3 years, but with renal replacement therapy, survival improves – up to more than 4 years. In the latter cases, infection is the major cause of death. With improved aggressive anti-infectious treatment, further enhanced survival is likely possible, even without specific treatment (10, 11).
Currently the focus of interest for research is pharmacological therapy to solubilize amyloid fibrils. Also research is currently underway to develop treatments that would increase tissue catabolism of amyloid deposits.
Footnotes
Financial support: None.
Conflict of interest: None.