
Abstract
Background
In the era of precision medicine, the suitability of fluoropyrimidine therapies in clinical oncology can be checked by pharmacogenetic investigations of single patients, thus optimizing resources and indicating the appropriate drugs to personalize their chemotherapy. For example, the presence of dihydropyrimidine dehydrogenase gene (DPYD) polymorphisms in cancer patients may lead to adverse effects when adopting fluoropyrimidine-based therapies.
Methods
We detected in a cancer patient a rare germline synonymous heterozygous variant of DPYD (c.1905C>T) in proximity to the exon 14 splice donor site. Because in silico analyses hypothesized potential deleterious effects of the splice site, we performed both quantitative and qualitative mRNA analyses to investigate the possible pathogenic nature of the variant.
Results
We did not detect any alterations in mRNA expression or in the cDNA sequence of DPYD gene transcript.
Conclusions
Our observations suggest that the c.1905C>T variant of DPYD does not have a pathogenic effect. Therefore, assessment of the clinical significance of rare sequence variants could emphasize the predictive value of DPYD gene alterations in identifying patients at potential risk for fluoropyrimidine-related toxicity.
Introduction
Dihydropyrimidine dehydrogenase (DPYD) (OMIM 612779) is the initial and rate-limiting enzyme in catabolism of both thymine and uracil pyrimidine bases of DNA and is essential for their degradation to beta-alanine and beta-isobutyric acid, respectively. Molecular analysis of the DPYD gene, which is located on chromosome band lp22 and includes 4,399 nucleotides and 23 coding exons, enables the identification of mutations that modify both expression and catalytic activity of the enzyme (1). DPYD deficiency (OMIM 274270) is a genetically determined condition with an autosomal dominant transmission that is characterized by enriched urinary excretion of both thymine and uracil in homozygous deficient patients, and is associated with variable clinical signs of psychomotor retardation, epilepsy and muscular hypertonia (2). Furthermore, this enzyme is involved in the catabolism of fluoropyrimidines, a class of drugs commonly used in cancer patients. In patients carrying both homozygous and heterozygous mutations, even in the absence of typical clinical signs associated with the altered metabolism of pyrimidines, administration of these chemotherapeutic agents may result in severe toxicities including neutropenia, thrombocytopenia, mucositis, hemorrhagic diarrhea and neuropathies (2).
Some sequence variants have been definitely associated with fluoropyrimidine-related toxicities (3). The most frequent variant of DPYD includes a point mutation in the splice donor site of the intron 14 (IVS14+1G>A; c.1905+1G>A; rs3918290; DPYD*2A) determining the exon 14 skipping (165 bp) and the synthesis of a nonfunctional protein causing severe fluoropyrimidine-induced toxicity (3). A less common mutation, associated with decreased DPYD activity and drug toxicity, is characterized by an amino acid substitution from a highly conserved Isoleucine to a Serine residue at the codon 560(Ile560Ser; 1679T>G; rs55886062; DPYD*13) (4, 5). Finally, the c.2846A>T variant leads to a structural change in the DPYD enzyme (Asp949Val; rs67376798) that interferes with cofactor binding and electron transport (3).
Therefore, for patients carrying these variants, a reduction of the fluoropyrimidine dose is recommended in case of heterozygosity, or the adoption of an alternative drug in cases of homozygosity, as suggested by the main scientific societies and working groups in this field (3, 6, 7).
In particular, the US Food and Drug Administration (FDA; http://www.fda.gov/, accessed 26 March 2017), the Dutch Pharmacogenetics Working Group and the European Medicines Agency (EMA; http://www.ema.europa.eu/ema, accessed 26 March 2017) recommend not to administer fluoropyrimidine-based therapies in patients with DPD enzyme deficiency (7). The National Comprehensive Cancer Network (NCCN) and the American Society of Clinical Oncology (ASCO) do not require the analysis of DPYD mutations before therapy but only in cases of severe adverse events during fluoropyrimidine therapy. According to the European Society for Medical Oncology (ESMO), the pharmacogenetic analysis of the DPYD gene in the pretherapy setting is an option but is not routinely recommended (7). The Clinical Pharmacogenetics Implementation Consortium (CPIC; https://cpicpgx.org/, accessed 26 March 2017) suggests an alternative drug for homozygous patients and a dose reduction at least 50% in carriers of heterozygous genotypes (6). The Associazione Italiana di Oncologia Medica (AIOM; the Italian Association of Medical Oncology) and the Società Italiana di Farmacologia (SIF; the Italian Society of Pharmacology) have adopted the same fluoropyrimidine dose adjustments as those of the CPIC, but the pharmacogenetic test is recommended before starting fluoropyrimidine in cases of a high risk to benefit balance (http://www.sifweb.org/docs/sif_aiom_position_paper_raccomand_farmacogen_gen15.pdf, accessed 26 March 2017).
Case report
A 75 year-old woman was referred to our oncogenomic laboratory for pharmacogenetic analyses before adjuvant chemotherapy after colectomy to remove an adenocarcinoma of intestinal type of the transverse colon (T4N2M1) infiltrating the cecum, the last ileal loop and a jejunal area. Molecular analysis of the neoplasia, performed in a different structure, showed a K-RAS c.351A>T; p.Lys117Asn (K117N) mutation. To identify potential risk of toxicity related to the chemotherapy, molecular analysis of the DPYD variants IVS14+1G>A, 1679T>G and 2846A>T was performed on genomic DNA isolated from peripheral blood using a QIAamp DNA Blood extraction kit (Qiagen Inc., Chatsworth, CA, USA). Sequencing reactions were performed using a Big Dye Terminator (Thermo Fisher Scientific Inc.) on a 3500 Series Genetic Analyzer (Thermo Fisher Scientific Inc.). To exclude preanalytical and analytical errors, PCR reactions and sequencing analyses were carried out on 2 different DNA extractions. For both primer design and results' control, we referred to Ensembl sequence DPYD-001 (ENST00000370192.7).
The comprehensive evaluation of the 3 mutations investigated in DPYD revealed a wild-type genotype while the electropherograms relative to exon 14 showed a heterozygous transition from C to T in the last nucleotide of the exon (c.1905C>T) that was not enough to cause an amino acid change (p.Asn635Asn) (Fig. 1).
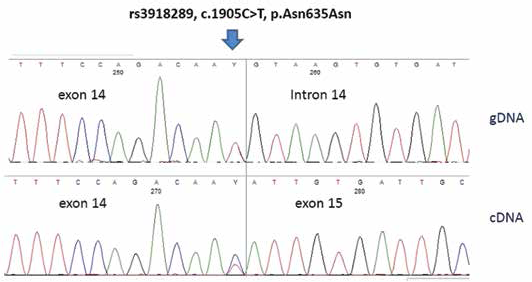
Direct sequence analysis of the DPYD c.1905C>T variant detected in our patient. The upper panel shows the gDNA sequence of exon 14 and intron–exon border. The lower panel shows the patient cDNA sequence, performed with primers spanning exons 13 and 15, demonstrating the integrity of exon 14.
The variant was recognized as the reference SNP (refSNP) Cluster Report rs3918289 (http://www.ncbi.nlm.nih.gov/snp/, accessed 26 March 2017), and described by the clinical variant database ClinVar (http://www.ncbi.nlm.nih.gov/clinvar/, accessed 26 march 2017; ID: 100087) as a polymorphism for which there is no clinical evidence of pathogenicity. To evaluate the impact of the proximity of the detected sequence variant to a donor site of splicing, we used the most popular bioinformatics Web tools to predict potential functional alterations of splicing sites. The prediction of a possible role of the variant on the adjacent splice donor site referred to a “potential alteration of splicing” due to an “alteration of an ESE (Exonic Splicing Enhancers) site,” according to the Human Splicing Finder website (http://www.umd.be/HSF3/index.html, accessed 26 March 2017) (Fig. 2). This site uses 12 different algorithms to predict the mutation effect on both acceptor and donor splice sites, including the evaluation of exon splicing enhancers (ESEs) and silencers (ESSs) (8). A similar prediction of the splice site alteration was also suggested by the RESCUE-ESE Web Server indicating the sequence variant to be responsible for the replacement of an ESE hexamer at the exon–intron junction (Fig. 2) (9), while other prediction websites, such as SplicePort (http://spliceport.cbcb.umd.edu/, accessed 26 March 2017), did not indicate any transcription abnormalities for this case.

Screenshots illustrating the in silico analyses, performed on wild-type and c.1905C>T variant patient sequences, using a Web tools predictor of potential functional alterations of splicing sites. The upper panel shows the results from the Human Splicing Finder website, a tool for studying pre-mRNA splicing, which uses 12 different algorithms to predict the potential effects of mutations on splicing motifs (http://www.umd.be/HSF3/technicaltips.html, accessed 26 March 2017). The lower panel shows the graphic result of the RESCUE-ESE website, able to identify 238 hexamers as exonic splicing enhancers (ESEs) candidates from a database of 30,000 human exon–intron structures (http://genes.mit.edu/burgelab/rescue-ese/, accessed 26 March 2017).
Furthermore, to better characterize the potential pathogenicity of the identified variant, we analyzed the transcripts of DPYD exons 13-15 through cDNA sequencing and evaluated the mRNA levels. The total RNA was extracted from fresh peripheral blood mononuclear cells (PBMCs) using an RNA Mini Kit (Qiagen) and reverse transcribed into cDNA with an iScript cDNA Synthesis Kit (BioRad). Complementary DNA PCR reactions and direct sequence analysis were performed using a new pair of primers spanning exons 13 and 15 of the DPYD gene. After mRNA retrotranscription and subsequent direct sequencing, no exon 14 skipping was found (Fig. 1).
A quantitative real-time PCR assay was performed in 3 replicates per sample, using the iTaq Universal SYBR Green Supermix (BioRad) in the Step One Plus instrument (Applied Biosystems). The mRNA level was measured with the comparative threshold cycle (Ct) method using glyceraldehyde-3-phosphate dehydrogenase (GAPDH) as endogenous control and relative values as calculated by ΔCt. The analysis was extended to 4 healthy subjects (from the personnel of our lab), who had previously been genotyped and identified as having the wild type for the 3 variants of the DPYD. The evaluation of mRNA levels in the patient carrying the variant was completely similar to those observed in the healthy subjects expressing the wild type of DPYD. On the basis of the clinical features and our analysis, the patient received bevacizumab plus capecitabine and oxaliplatin every 2 weeks for 6 cycles without any evidence of fluoropyrimidine-related toxicity.
Conclusions
Here, we describe a rare variant of the DPYD gene (c.1905C>T) in a patient with colon cancer. At the protein level, this variant, localized at the last nucleotide of the DPYD exon 14, does not result in any amino acid substitution. This polymorphism was previously classified as of unknown clinical effect in the ClinVar database (ID: 100087). Therefore, to assess its hypothetical pathogenic role, we used several Web tools to identify any potential deleterious effects of the protein. The prediction of its pathogenicity by several websites led us to perform both qualitative and quantitative mRNA analyses, which in all instances revealed a nondeleterious effect for this variant. We were unable to perform functional enzymatic tests, but given the synonymy of the identified mutation and the complete concordance in mRNA assays, we reasonably considered this polymorphism as nonpathogenic for fluoropyrimidine-related toxicities, and with no functional significance for DPYD enzyme activity (3).
Fluoropyrimidines are commonly utilized anticancer drugs. The increasing application of pharmacogenetic assays in clinical oncology devoted to emphasizing personalized medicine, allows the discovery of new rare mutations in the DPYD gene that may also account for an appreciable percentage of the fluoropyrimidine toxicity (10). Developments in pharmacogenomic research often take advantage of the identification of rare but functional polymorphisms that can offer an important contribution to predicting drug responses in malignancies. As suggested by recent studies, the use of in silico and in vitro functional assays may lead to the characterization of many new variants of as-yet unknown significance that are progressively being identified in mutational analyses, enabling a precise genotype–phenotype correlation (11). Therefore, the identification and characterization of potential pathogenic sequence variants are relevant for personalized dose adjustments to fluoropyrimidine in clinical practice (3, 10). The rare polymorphism of DPYD gene identified in our patient may thus help to update specific databases and suggest to clinicians that its detection is not predictive of risk for fluoropyrimidine-related toxicities.
Footnotes
Financial support: This work was partially supported by a grant from the Italian Association for Cancer Research (AIRC, IG11647) (to F. S.) and from the Apulia Region (Oncogenomic Project).
Conflict of interest: The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in, or financial conflict with, the subject matter or materials discussed in the manuscript apart from those disclosed.