
Abstract
Background:
Many cancer patients who receive chemotherapy experience adverse drug effects. Pharmacogenomics (PGx) has promise to personalize chemotherapy drug dosing to maximize efficacy and safety. Fluoropyrimidines and irinotecan have well-known germline PGx associations. At our institution, we have delivered PGx clinical decision support (CDS) based on preemptively obtained genotyping results for a large number of non-oncology medications since 2012, but have not previously evaluated the utility of this strategy for patients initiating anti-cancer regimens. We hypothesize that providing oncologists with preemptive germline PGx information along with CDS will enable individualized dosing decisions and result in improved patient outcomes.
Methods:
Patients with oncologic malignancies for whom fluoropyrimidine and/or irinotecan-inclusive therapy is being planned will be enrolled and randomly assigned to PGx and control arms. Patients will be genotyped in a clinical laboratory across panels that include actionable variants in UGT1A1 and DPYD. For PGx arm patients, treating providers will be given access to the patient-specific PGx results with CDS prior to treatment initiation. In the control arm, genotyping will be deferred, and dosing will occur as per usual care. Co-primary endpoints are dose intensity deviation rate (the proportion of patients receiving dose modifications during the first treatment cycle), and grade ⩾3 treatment-related toxicities throughout the treatment course. Additional study endpoints will include cumulative drug dose intensity, progression-free survival, dosing of additional PGx supportive medications, and patient-reported quality of life and understanding of PGx.
Discussion:
Providing a platform of integrated germline PGx information may promote personalized chemotherapy dosing decisions and establish a new model of care to optimize oncology treatment planning.
Background
The vast majority of patients who receive chemotherapy report side effects, many of which are grade 3 or higher toxicities requiring medical interventions.1 –3 Pharmacogenomics (PGx) – the study of how germline genetic variants affect individual response to medications – has promise to personalize drug dosing to optimize safety and efficacy. Many oncologic therapeutics have well-known PGx associations, and preemptive genotyping and the use of germline PGx information offers an opportunity to improve oncology care by identifying individuals at risk of adverse drug effects (ADEs).4 –8 However, aside from thiopurine methyltransferase (TPMT) testing prior to 6-mercaptopurine administration in pediatric oncology, PGx has not been routinely incorporated into oncologic practice in the vast majority of countries (including the US), and in fact, oncology may lag behind other fields.9,10 This is somewhat surprising, as oncologists routinely utilize information on germline cancer predisposition and somatic (tumor-based) genomic alterations for patient care planning (notably in the context of Poly ADP ribose polymerase (PARP) inhibitors, in which BRCA germline variants are hypothesized to accompany somatic susceptibility to therapy).11,12 The potential application of germline PGx information offers an additional avenue for the delivery of personalized medicine in oncology.13,14
Fluoropyrimidines (5-fluorouracil [5-FU], capecitabine) and irinotecan are commonly prescribed chemotherapies, with efficacy across a broad range of tumor types. Dihydropyrimidine dehydrogenase (DPD) is the rate-limiting enzyme in fluoropyrimidine metabolism. 15 Polymorphisms in the DPYD gene can result in enzyme deficiency leading to an increased risk of severe, sometimes fatal toxicities in up to 10% of patients. 8 Allele frequencies of these variants vary among ethnic groups and confer different levels of predicted enzymatic activity, also called ‘activity scores’. Table 1 shows several well-described clinically actionable alleles in DPYD, with their associated enzymatic activity scores and observed frequencies in various populations.
Selected actionable DPYD variants, with associated functional activity scores and expected frequencies by ethnicity.

Enzyme activity is defined as 1 = normal function, 0.5 = reduced function, 0 = no function. An individual’s activity score is defined as the sum of the two diploid alleles’ variant activity scores.
Caucasian ancestry cohort includes European and European-descent North American populations.
Other ancestry cohort includes Asian (East, South), Indian, Middle Eastern, and Americas populations.
The feasibility and safety of fluoropyrimidine genotype-guided dosing has now been demonstrated by several groups.16,17,22 –25 Deenen et al. prospectively screened patients initiating fluoropyrimidine therapy for DPYD*2A; among >2000 patients screened, 22 patients were identified as *2A heterozygotes, and 18 preemptively received reduced fluoropyrimidine dosing (the other four did not receive a fluoropyrimidine). 16 The incidence of severe toxicities (grade ⩾3) among carriers was 28%, which compared very favorably with the observed toxicity of a historical cohort of patients with DYPD*2A variants who received full-dose fluoropyrimidines (grade ⩾3 toxicity rate 78%), and with the concurrently treated group of patients who did not carry *2A and received full dose fluoropyrimidines (23% grade ⩾3 toxicity rate). Compared to the historical cohort of *2A carriers who received full dosing, the risk of drug-induced death was reduced from 10% to 0% in *2A carriers receiving reduced dosing. Furthermore, the average total treatment cost was modeled as being modestly lower with screening, even when including the screening costs for the entire population. Subsequently, Henricks et al. prospectively evaluated genotype-guided dosing that was extended to include four DPYD variants (*2A or c.1905+1G>A, c.2846A>T, *13 or c.1679T>G, and HapB3/c.1236G>A). 17 Heterozygous variant allele carriers comprised 8% of all patients evaluated (85 of 1103 patients) and received initial chemotherapy dose reductions. The relative risk of severe fluoropyrimidine-related toxicity was reduced in all groups as compared to historical controls. Based on these and other data, the Clinical Pharmacogenetics Implementation Consortium (CPIC) recommends upfront dose reductions for carriers of these alleles. 18 Finally, recent data have also demonstrated that the DPYD variant c.557A>G (rs115232898, p.Y186C), which is present in 3–5% of individuals with African ancestry, results in reduced functional DPD enzyme levels and is likewise associated with higher rates of fluoropyrimidine toxicity.18,20 A number of studies (both observational and prospective) are ongoing to evaluate the impact of DPYD variant testing in cancer patients, albeit in a non-randomized fashion.26 –28
Similarly, the active metabolite of irinotecan, 7-Ethyl-10-hydroxycamptothecin (SN-38), is glucuronidated (inactivated) by the enzyme uridine diphosphate (UDP) glucuronosyltransferase family polypeptide A1, which is encoded by the UGT1A1 gene. 29 The UGT1A1 gene has common thymine-adenine (TA) insertion/deletion polymorphisms in the promoter region, and some populations also manifest a missense variant in exon 1, either of which results in an altered risk of treatment-related toxicities, especially neutropenia.30 –33 Table 2 shows several well-described clinically actionable alleles in UGT1A1, with observed frequencies in various populations. The wild-type allele (UGT1A1*1) has six TA repeats, while the most common variant allele in Caucasians (UGT1A1*28) has seven TA repeats. Approximately 10% of European-descent populations are homozygous for the UGT1A1*28 allele (TA indel/UGT1A1 7/7), while an additional 40% are heterozygotes for this variant. The greater number of promoter region repeats has been shown to result in less transcription; thus, patients – particularly those who are homozygous for the UGT1A1*28 polymorphism (or the *37 allele which has eight TA repeats and is associated with African ancestry) have reduced SN-38 clearance and are at higher risk of hematologic toxicity including severe neutropenia as well as dose-dependent severe diarrhea when receiving irinotecan.5,6,30,32 –34 Patients with the UGT1A1*36 allele have five TA repeats and are not at increased risk of toxicities. 35 In addition to TA repeat variants, patients carrying the UGT1A1*6 (or 211G>A) polymorphism in Exon 1 of the gene (most commonly observed in individuals of Asian ancestry) also have reduced enzyme activity and SN-38 clearance, which is also associated with an increased risk of irinotecan-related toxicities including severe neutropenia and diarrhea.36 –42 The Food and Drug Administration (FDA) product label in the United States recommends that a lower starting dose of irinotecan should be considered for patients with the UGT1A1*28/*28 genotype. 43
Selected actionable UGT1A1 variants, with associated functional implications and expected frequencies by ethnicity.

Caucasian ancestry cohort includes European and European-descent North American populations.
Other ancestry cohort includes Asian (East, South), Indian, Middle Eastern, and Americas populations.
Despite a relatively large body of evidence demonstrating the feasibility, safety, and cost-effectiveness of PGx testing for DPYD and UGT1A1, and despite FDA label prescribing information regarding relevant PGx for each, prospective genotyping for DPYD and UGT1A1 prior to the administration of fluoropyrimidines and irinotecan is essentially non-existent in the United States.16,17,33,47 For fluoropyrimidines, only very recently did the European Medicines Agency recommend routine upfront testing of DPD deficiency. 48 We hypothesize that the lack of routine PGx testing in the United States is due to a perceived deficiency of prospective, randomized data, as well as a paucity of appropriate systems for obtaining and translating germline PGx information to oncology clinicians at the bedside.49,50 We seek to address these implementation barriers, as well as this important evidence gap, with the design of the prospective, randomized PhOCus Trial: Implementation of Pharmacogenomic Testing in Oncology Care.
Methods
Design
This is a randomized, prospective study to evaluate the effects of PGx testing on chemotherapy dosing decisions and on reducing medication-related adverse events in oncology patients. Patients with cancer who are planned to receive fluoropyrimidine (5-FU, capecitabine) and/or irinotecan therapy will be enrolled and randomly allocated to PGx and control arms (Figure 1). In the PGx arm, providers will be given immediate access to patient-specific information and genotypic dosing guidance (based on patient DPYD and UGT1A1 variant allele status) in the form of the electronic medical record (EMR)-integrated software tool, the Genomic Prescribing System (GPS). In the control arm, initial fluoropyrimidine and irinotecan dosing will occur as per standard of care, and subject genotyping will be deferred until approximately 6 months after enrollment (after the course of chemotherapy).

The ‘PhOCus Trial’ study design.
The co-primary endpoints are dose intensity deviation rate (the proportion of patients receiving dose modifications) during the first treatment cycle, and the incidence of grade 3 or higher toxicities throughout the entire treatment course. Secondary endpoints will include overall chemotherapy dose intensity and key oncologic efficacy endpoints including response rate, progression-free survival (PFS), and overall survival (OS). Exploratory endpoints will include the impact of PGx on prescribing of additional PGx-informed oncology-related and supportive care medications, patient-reported quality of life, and patient understanding of PGx. We hypothesize that if oncology clinicians are provided preemptive PGx information to help guide fluoropyrimidine and irinotecan dosing, they will dose-modify treatments in an effort to mitigate and avoid toxicities, which may improve treatment tolerability as well as outcomes for cancer patients.
Subjects
Adult oncology patients (18 years or more) receiving care at the University of Chicago Comprehensive Cancer Center, and for whom treatment with a fluoropyrimidine and/or irinotecan treatment-containing regimen is being considered, are eligible. Specifically, enrollment will occur in breast, gastrointestinal (GI), and head and neck medical oncology clinics, given the common utilization of these agents in standard of care treatment. Recruitment will occur from across each of two physical cancer center locations, including the university campus main medical center in Chicago, and a university-affiliated community-based oncology practice network site in suburban Chicago. Patients will be treatment-naive for the planned chemotherapy agent of interest. Subjects will be approached for enrollment by the research coordinator in conjunction with standard of care oncology visits. Exclusion criteria include: (a) prior exposure to the planned chemotherapy of interest (fluoropyrimidine and/or irinotecan); (b) enrollment in another investigational trial which would preclude dose modifications of fluoropyrimidine and/or irinotecan chemotherapies; (c) history of or active consideration for bone marrow, liver, or kidney transplantation; (d) history of or active blood cancer (e.g. leukemia); (e) chronic kidney disease, as defined by estimated glomerular filtration rate (eGFR) <30/mL/min/1.73 m 2 , due to the risk of decreased drug excretion; and (f) liver dysfunction, as defined by the following laboratory values, due to the risk of decreased drug metabolism: total bilirubin ⩾1.5 mg/dL, aspartate aminotransferase (AST) and alanine aminotransferase (ALT) ⩾ 2.5 × upper limit of normal (AST and ALT ⩾ 5 × upper limit of normal if hepatic metastases are present).
Genotyping
At enrollment, all patients will provide a blood sample for PGx germline genotyping. Genotyping will be carried out in a College of American Pathologists (CAP)-accredited and Clinical Laboratory Improvement Amendments (CLIA)-certified laboratory at the University of Chicago. The anticipated test turnaround time is 7–10 business days. All patients in the study will give consent to genotyping across our established custom panel of germline variants identified as affecting drug disposition, response, or toxicity (using OpenArray technology from Thermofisher). 51 Patients will be genotyped specifically for five actionable variants within DPYD (Table 1). In an exploratory fashion, patients will also be genotyped for several previously implicated variants in the genes encoding for thymidylate synthase and thymidylate phosphorylase, although the genotype results for these variants will not be delivered clinically.52,53 For UGT1A1, the UGT1A1*6 exon variant will be assessed by OpenArray, while promoter sequence thymine-adenine-thymine-adenine (TATA) polymorphisms (UGT1A1*28, *36 and *37) will be assessed by a separate assay using polymerase chain reaction (PCR) and sizing by polyacrylamide gel (Table 2). 54 To guide the potential use of supportive care medications (exploratory endpoint) separately, we will also genotype patients across our custom dedicated CYP2D6 panel, which uses the Invader technology from Hologic, complemented by CYP2D6 copy number assessment.55,56
Translation of genotype results and clinical decision support via the genomic prescribing system
Multiple genomic profiling assays based on next-generation sequencing (NGS) are utilized routinely in oncologic patient care planning to assess for somatic (tumor-based) genomic alterations, providing patient-specific results along with interpretations and recommendations to help guide personalized treatment decisions. However, there is a lack of implementation of germline PGx testing in oncology, potentially due to a scarcity of appropriate systems for obtaining and translating this information. Our institution has previously employed the secure, password-protected, PGx results and decision support portal, the GPS, an interactive software tool that is linked to our institutional EMR51,57 (Figure 2). After genotyping, oncology clinicians (physicians, advanced practice providers and nurses), upon opening their patient’s chart, will receive a best practice alert (BPA) within the EMR notifying them of the PGx results with a direct link to the GPS, which will then deliver information as a drug-centered ‘drug–gene pair result summary’. This summary will contain a synopsis of the patient’s genotyping results, an interpretation, prescribing recommendations, as well as literature references about the drug–gene pair to help guide personalized prescribing. In addition, should a subject’s genotyping results determine that they are at high risk of developing ADEs (such as in the case of DPYD homozygosity), clinicians will be directly contacted by email by the study team. Tables 3 and 4 detail the specific chemotherapy dosing recommendations (supported by and harmonized with international guidelines) that will be delivered to clinicians in this study, based on a patient’s DPYD and/or UGT1A1 variant allele status.

Pharmacogenomic clinical decision support provided via electronic medical record (EMR) embedded genomic prescribing system (GPS).
Recommended dosing modifications according to DPD metabolizer status.

Recommended dosing modifications according to UGT1A1 metabolizer status.

Assessments
In addition to general demographic and clinical data, a research database will be created, which will record identifying information for each subject, including tumor type and stage, age, gender, height, weight, date of sample collection, and PGx results. Subjects’ medical records will be reviewed by research staff on a continual basis at 8–12 week time points while enrolled, and the database will be populated to include:
Treatment
Patients will be randomly allocated to the following groups:
- PGx arm: Providers will be given access to patient-specific information and genotypic dosing guidance (based on patient DPYD and UGT1A1 variant allele status) in the form of the EMR-integrated software tool, the GPS, to inform initial chemotherapy dosing.
- Control arm: Initial fluoropyrimidine and irinotecan dosing will occur as per standard of care, and subject genotyping will be deferred (to occur approximately 6 months after enrollment; i.e. after the course of chemotherapy).
Endpoints
The co-primary endpoints are: (a) dose intensity deviation rate (the proportion of patients receiving modifications) during the first treatment cycle; and (b) the incidence of grade 3 or higher toxicities throughout the entire treatment course. Secondary endpoints will include: (a) overall chemotherapy dose intensity; and (b) progression-free survival (plus response rate and overall survival, as available). Exploratory endpoints will include: (a) the impact of PGx on prescribing of additional PGx-informed oncology related and supportive care medications; (b) patient-reported quality of life (QOL); and (c) patient understanding of PGx.
Chemotherapy dose determination
Each dose and date of chemotherapy administration will be recorded. Standard of care dosing will be defined by the signed chemotherapy plan, in which the dose is specified by the University of Chicago Chemotherapy Pharmacy Improvement Team based on tumor-specific National Comprehensive Care Network (NCCN) guidelines. Chemotherapy doses that deviate from this standard dosing will be calculated and compared between patients in both the PGx and control arms. The reasons for dose deviation, if available, will be documented.
Chemotherapy toxicity assessment
While the subject remains enrolled, clinicians will be asked to document toxicity occurrences, graded according to Common Terminology Criteria for Adverse Events (CTCAE) v5.0 64 in each progress note. Only ADEs thought attributable [with attribution score of 3 (possible), 4 (probable), or 5 (definite)] to fluoropyrimidine and/or irinotecan administration will be included in the primary endpoint analysis of the incidence of ⩾ grade 3 toxicities. For subjects receiving fluoropyrimidines, the following ADEs will be attributed as likely related to this agent’s administration (in the absence of another obvious cause) and graded as per Supplemental Table 1: neutropenia, diarrhea, hand-foot syndrome and mucositis (n.b.: the latter will be excluded as a primary fluoropyrimidine-related ADE in head and neck cancer patients receiving concomitant radiation therapy). For subjects receiving irinotecan, the following ADEs will be attributed as likely related to this agent’s administration (in the absence of another obvious cause) and graded as per Supplemental Table 2: neutropenia and diarrhea. Any ambiguous grade 3 or higher toxicity possibly related to either agent will be adjudicated by a panel of study reviewers who are blinded to treatment arm. Adverse events related to other chemotherapy or anti-cancer agents, from radiation therapy or from the underlying disease process, will be recorded in the database but will not be attributed or included in the primary analysis. Toxicity assessments will occur weekly during cycle 1 of chemotherapy, and on an ongoing basis at intervals of at least every 8–12 weeks while the subject remains on study. At the end of the study, a composite toxicity rate of ⩾ grade 3 adverse events will be calculated for both PGx and control arms.
Longitudinal survey results
At routine clinic visits with their oncology provider, we will assess subject views about the visit and about any treatment decisions that were made. These assessments will occur through the administration of a questionnaire by research staff at or immediately after the first visit, and at an interval of at least every 8–12 weeks while the subject remains enrolled. The questionnaires will query QOL, treatment decision-making, patient–provider interactions, the provider–patient relationship, and subject satisfaction with care to determine if there are differences between the two groups in these measures.57,65 –67
Standard protocol approval, registration, and patient consent
The project was approved by both the Clinical Trials Review Committee (CTRC) and the institutional review board (IRB) at the University of Chicago Medical Center as of 14 August 2020. It is registered at ClinicalTrials.gov (#NCT04541381). Every enrolled patient will provide written informed consent. Patients may withdraw from the study at any time without affecting their current or future care.
Duration of participant follow-up and subject withdrawal
Enrolled subjects will be followed for the duration of time they remain on fluoropyrimidine and/or irinotecan therapy, and for at least 1 month following chemotherapy completion to document treatment-related adverse events. Efficacy endpoints beyond this dedicated observation period will also be captured (e.g. PFS/OS). Subjects on maintenance chemotherapy will be followed for toxicity endpoints up to 6 months. Subjects may withdraw from the study at any time, and participation may be discontinued if they are lost to follow-up, the study is terminated, or the investigator feels that it is no longer in the subject’s best interest to participate.
Statistics
Upon enrollment subjects will be randomly allocated to an upfront genotyping or ‘PGx-guided’ arm versus a control arm. Timing of genotyping is determined by randomized assignment to either of these two groups. Randomization schema will be stratified by cancer type (breast, GI, head and neck), disease-specific stage, and treatment setting (adjuvant vs. metastatic, including line of metastatic therapy within this latter setting). Given institutional cancer-type volumes and strategic enrollment plans, we anticipate that half of all enrolled subjects will have GI malignancies, with the rest composed of patients with breast and head/neck malignancies. Based on prior institutional census data, we estimate that 75% of enrolled patients will be Caucasian (European/North American ancestry), and 25% will be of African ancestry. The study was powered based on the expected allele frequencies of DPYD and UGT1A1 variants displayed in Tables 1 and 2, with an estimated composite prevalence of actionable variants by ethnicity expected to be 19.2% and 19.1% for African and European ethnicities, respectively. For half of all subjects enrolled (i.e. those with GI malignancies) PGx information related to both fluoropyrimidines and irinotecan are of interest. For the other half of the subjects with head/neck or breast malignancies, we anticipate only fluoropyrimidine-related PGx information will be clinically relevant, and dedicated UGT1A1 testing will not be performed in these subjects. For these subjects, the estimated prevalence of variant alleles was based on DPYD only and is 4.7% and 3.2%, for European-descent and African-descent populations, respectively. Based on the above expected subject demographics and our disease clinic/cancer type enrollment plans, we estimate that for enrolled subjects of African ancestry, 11.2% of evaluable patients will carry an actionable variant affecting a chemotherapy of interest that they will receive. For subjects of European ancestry, a similar estimated 11.9% of treated patients will have an actionable variant. Thus, the study is conservatively powered for an approximate 10% actionable variant allele prevalence across all enrolled subjects.
In the first stage of the study, enrolled patients will be randomly assigned to PGx versus control arms in a 3:1 ratio to enrich the PGx arm for analysis of co-primary endpoint 1 (Figure 3). For co-primary endpoint 1, the dose intensity deviation rate (Pdev) (defined as the proportion of patients receiving cycle 1 dose modifications) will be measured for both arms. We assume dose intensity deviation rates of 10% in the control arm due to other clinical factors (e.g. fragility, performance status), an estimate based on historical evidence.68,69 For the PGx arm, we expect most clinicians proactively to dose reduce patients carrying risk alleles; thus, we conservatively estimate 50% dose modification rates (i.e. 50% concordance with PGx guidance) in individuals with at-risk variant alleles. Stage 1 randomization will be performed in a 3:1 ratio so that analysis is powered to detect a dose deviation difference, Pdev. Dosing deviation (Pdev) will be calculated and compared between patients with risk alleles in the PGx arm versus all patients in the control arm for the first cycle of chemotherapy. As a secondary analysis, we will examine whether clinicians’ decisions on dose modification correlates with any of the demographic or clinical variables of interest, including age, gender, ethnicity, cancer type or stage, and at-risk allele status.
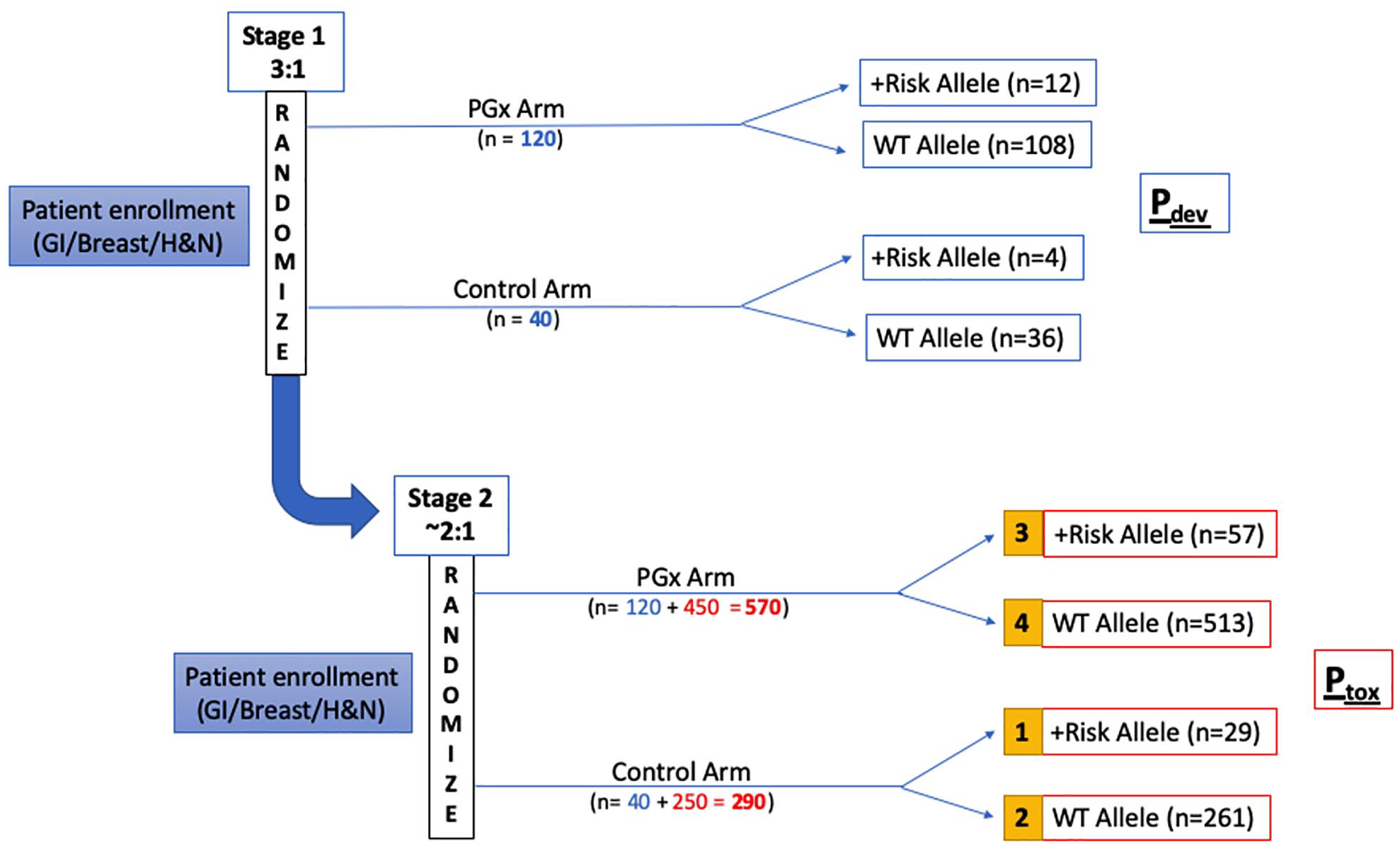
The ‘PhOCus Trial’ group assignment and analysis plan.
After the accrual of 160 patients (120 in the PGx arm, 40 in the control arm; defined as the end of stage 1), enrollment will be halted, and an interim analysis will be performed to confirm that the expected variant allele frequency rates have been observed. If no meaningful difference is detected between the observed and expected frequencies, then the risk considerations will be deemed not to have changed for subsequent subjects, and the study will proceed to enrollment of the remaining patients.
In stage 2, patients will be randomly assigned to PGx vs. control arms in a 1.8:1 ratio, for evaluation of the second co-primary endpoint, Ptox (toxicity rate of grade 3 or higher adverse events). The sample size calculation for co-primary endpoint 2 of the study is based on the expected incidence of grade 3 or higher toxicity rate among patients with the at-risk allele versus wild-type (WT), as established in prior literature (Ptox of 70% and 30%, respectively). 16 The primary analysis will be performed comparing toxicity rates between group 1 (variant allele carriers in control arm) and group 3 (variant allele carriers in PGx arm). Logistic regression analysis will also be performed to evaluate the association between the incidence of adverse events with variables of interest such as age, gender, ethnicity, cancer type, at-risk allele status, study arm, and chemotherapy dosing intensity (standard of care or modified dosing). If patients are genotyped via alternative means, or in the case that genotyping results are not available at the time of treatment initiation, patients will be analyzed on an intent-to-treat basis.
Secondary endpoints will be analyzed as follows:
At the conclusion of the study, the cumulative chemotherapy drug dose intensity (the function of dose and frequency of drug administration) received by each subject during the entire treatment course will be calculated.
Response and survival assessments will be analyzed, including response rate, PFS, and OS. These endpoints will be analyzed by tumor type, stage, and disease setting, and will be compared between arms and within each arm compared to historical controls treated per standard of care.
Exploratory endpoints will be analyzed as follows:
Many medications commonly utilized in oncologic patients have well established PGx guidance and have been included in the GPS. Patient-specific information and dosing recommendations for these medications will be available to clinicians along with their genotyping results through GPS access.
Subject-reported QOL and understanding of PGx will be captured by survey instruments administered to subjects in both arms at enrollment, weekly during cycle 1, and on an ongoing basis of 8–12 week intervals while the patient remains on study.
Potential benefit for participants
All participants will receive PGx testing that could aid their clinicians in prescribing decisions for chemotherapy, and other PGx informed medications. This could potentially permit the avoidance of medications, or doses, which might be harmful to the subject, or alternatively, it might allow the identification of subjects as particularly likely to benefit from a given drug or therapy.
Potential risks and burdens for participants
The primary potential risks of participation are those associated with potential adverse outcomes to subjects if providers make medication or chemotherapy dosing changes that are based on subject-specific PGx findings that result in harm and/or altered anti-tumor efficacy; however, anti-tumor efficacy is thought to be preserved given prior literature demonstrating similar toxicity rates and pharmacokinetic parameters in patients carrying variant alleles receiving dose modifications.16,17,33,47,54,60,70
Secondly, only select variants of DPYD and UGT1A1 (Tables 1 and 2) for which published studies demonstrating their PGx relevance will be tested. Chemotherapy drug metabolism, efficacy, and risk of toxicity may be affected by additional genetic factors that will not be evaluated. Thus, despite preemptive dose modifications, patients may develop toxicities due to other factors.
Conclusion
Chemotherapies such as fluoropyrimidines and irinotecan have well-known germline PGx associations. Preemptive genotyping offers the potential to identify individuals at increased risk of ADEs and improve patient outcomes. Our project seeks to demonstrate the potential benefit of utilizing preemptive PGx testing to provide individualized chemotherapy dosing. To our knowledge, this study will be the first of its kind to implement broad preemptive PGx information, in a prospective randomized fashion, across the oncology care setting (that is, for multiple malignancies). Providing this platform of integrated germline PGx information may enable personalized chemotherapy dosing decisions and establish a new model of care, incorporating comprehensive genomic data to optimize oncology treatment planning. This may improve tolerability and outcomes for cancer patients receiving commonly prescribed chemotherapies.
Supplemental Material
sj-pdf-1-tam-10.1177_1758835920974118 – Supplemental material for Implementation of pharmacogenomic testing in oncology care (PhOCus): study protocol of a pragmatic, randomized clinical trial
Supplemental material, sj-pdf-1-tam-10.1177_1758835920974118 for Implementation of pharmacogenomic testing in oncology care (PhOCus): study protocol of a pragmatic, randomized clinical trial by Natalie Reizine, Everett E. Vokes, Ping Liu, Tien M. Truong, Rita Nanda, Gini F. Fleming, Daniel V.T. Catenacci, Alexander T. Pearson, Sandeep Parsad, Keith Danahey, Xander M. R. van Wijk, Kiang-Teck J. Yeo, Mark J. Ratain and Peter H. O’Donnell in Therapeutic Advances in Medical Oncology
Footnotes
Conflict of interest statement
M.J. Ratain is a coinventor holding patents related to pharmacogenetic diagnostics and receives royalties related to UGT1A1 genotyping.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the University of Chicago Committee on Clinical Pharmacology and Pharmacogenomics T32 training grant NIH 5T32GM007019-41 (N. Reizine as a trainee) and the Benjamin McAllister Research Fellowship Award (N. Reizine as a trainee).
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.