
Abstract
Decreased dihydropyrimidine dehydrogenase enzyme activity is associated with severe fluoropyrimidine-associated toxicity. Four clinically relevant variants in the DPYD gene are associated with decreased dihydropyrimidine dehydrogenase activity. However, only ∼25% of DPYD variant carriers show a decreased dihydropyrimidine dehydrogenase activity in peripheral blood mononuclear cells.
Objective
To investigate if dihydropyrimidine dehydrogenase phenotyping has added value when combined with DPYD genotyping in predicting fluoropyrimidine-related toxicity.
Methods
Retrospective cohort study in which treatment and toxicity data were collected of 228 patients genotyped for four DPYD variants and phenotyped using an ex vivo peripheral blood mononuclear cell assay.
Results
Severe toxicity occurred in 25% of patients with a variant and normal dihydropyrimidine dehydrogenase activity, in 21% of patients without a variant and with decreased dihydropyrimidine dehydrogenase activity, and in 29% of patients without a variant and with normal dihydropyrimidine dehydrogenase activity (controls). The majority of patients with a variant or a decreased dihydropyrimidine dehydrogenase activity received an initial dose reduction (68% and 53% vs 19% in controls) and had a lower mean dose intensity (75% and 81% vs 91% in controls). Fifty percent of patients with a variant and decreased enzyme activity experienced severe toxicity, despite the lowest initial dose and whole treatment dose intensity. They also experienced more grade 4/5 toxicities.
Conclusions
Our results indicate that a combined genotype–phenotype approach could be useful to identify patients at increased risk for fluoropyrimidine-associated toxicity (e.g. patients with a variant and decreased dihydropyrimidine dehydrogenase activity). Because the group sizes are too small to demonstrate statistically significant differences, this warrants further research in a prospective study in a larger cohort.
Keywords
Introduction
Fluoropyrimidines (fluorouracil (5-FU) and its prodrug capecitabine) are commonly used in the treatment of several types of malignancies.1–3 Previous studies have shown that non-individualized fluoropyrimidine treatment causes severe toxicity in 6–34% of patients (grade ≥3, based on the Common Terminology Criteria for Adverse Effects (CTCAE) grading-system).4–7 Toxic effects of fluoropyrimidines mainly include stomatitis, mucositis, myelosuppression, and hand-foot syndrome. Many different factors including age, sex, renal function, liver function, co-medication, and other environmental factors are thought to influence the risk of toxicity.8,9
Over 80% of the active 5-fluorouracil (5-FU) is metabolized in the liver. 10 The most important enzyme in the metabolism of fluoropyrimidines is dihydropyrimidine dehydrogenase (DPD). 11 At least 3–5% of the Caucasian population has a decreased or absent DPD enzyme activity.12,13 As these findings strongly depend on methods used to measure DPD activity, considerably higher numbers of patients with decreased DPD activity have also been reported. 14 Several studies have shown a link between a decreased DPD enzyme activity and severe fluoropyrimidine toxicity. Between 36 and 59% of the patients who experience severe fluoropyrimidine toxicity (grade ≥3) have a decreased DPD enzyme activity.15–17 Only in a minority of individuals, the reduced DPD activity can be explained by genetic variants in the DPYD gene.14,18,19
The European Medicines Agency recommends DPYD genotyping prior to fluoropyrimidine treatment after publication of a prospective trial, demonstrating that DPYD genotype-based dose reductions improved patient safety during fluoropyrimidine treatment. 20 The four clinically relevant genetic variants in the DPYD gene that are known to be associated with a decreased DPD activity are DPYD*2A, DPYD*13, c. 2846A>T, and c.1129-5923C>G.18,19,21 The allele frequency of these variants is relatively low in the Caucasian population (between 0.03% and 1.35%). 22 The gene activity score (GAS) is often used to translate the DPYD genotype into a DPD phenotype to describe DPD activity and for dose recommendations. 23 This translation of DPYD genotypes to their functional activity is based on in vitro and in vivo studies and the subsequent GAS ranges from 0 (no DPD activity) to 2 (normal DPD activity). For heterozygous carriers of any of the clinically relevant variants, the Dutch Pharmacogenetics Working Group (DPWG) guidelines now recommend a dose reduction of 50%,24,25 instead of 50% for variants with a GAS of 1.0 and 25% for a GAS of 1.5 as was recommended before. 23 This change in dose recommendation is supported by the Clinical Pharmacogenetics Implementation Consortium (CPIC) in the most recent update of their guideline for fluoropyrimidines and DPYD. 26 Other rare and common DPYD genetic variants have been reported but are not included in the current international dosing guidelines due to insufficient evidence for functionality.18,27 When it is not possible to calculate the GAS based on DPYD genotype, for example when two variants might be located on the same allele, the DPWG recommends to determine DPD activity and adjust the initial dose based on available data.
DPYD genotyping is widely used globally. However, DPD enzyme activity has been underexplored. We previously showed that only 25% of patients with a decreased DPD enzyme activity carries one of the four DPYD variants. 18 So although DPYD genotyping (of four variants) has proven to be useful in preventing toxicity and is cost-effective,28,29 the majority of the patients with a reduced enzyme activity might remain unidentified and are possibly susceptible to severe fluoropyrimidine toxicity.
So far, one trial describing a combined genotype–phenotype approach has been published. 14 In this prospective trial, the occurrence of severe toxicity (early grade 4–5 toxic events) was compared between two groups. The first group was screened for DPD deficiency using a multi-parametric approach: determining uracil and dihydrouracil concentrations in blood, along with demographic characteristics, and DPYD genotyping. In the second group no pre-treatment testing was performed. The authors conclude that multiparametric pre-therapeutic DPD deficiency screening significantly avoided early severe life threatening toxic events. To our knowledge, no publications exist investigating the added value of the combined genotype–phenotype approach compared to DPYD genotyping alone, which is already widely implemented. Therefore, the aim of our retrospective study was to describe the value of combined DPYD genotyping and DPD phenotyping in predicting fluoropyrimidine-related toxicity in an existing hospital population, reflecting clinical practice.
Methods
Study design and group characteristics
This retrospective cohort study was performed in a single academic hospital in the Netherlands. It was approved by the local medical ethical committee. All 228 participants in this study were adults (≥18 years) diagnosed with cancer that were treated with fluoropyrimidines, either with capecitabine or 5-FU intravenously. The patients in this study were of different ethnic backgrounds, although the majority was Caucasian. Details on ethnicities will not be discussed because this was poorly reported. Fluoropyrimidine treatment was only given when the treating physician assessed the patient's condition to be sufficient, based on clinical presentation and laboratory parameters. Previous systemic therapy was allowed with resolved adverse events to grade ≤1. The patients received treatment in the period from January 2014 to December 2019. In case patients received more than one fluoropyrimidine treatment line in this period, all treatments were included in the analysis. This patient cohort is partly overlapping with the cohort described in a previous study which investigated the relationship between genetic variants in DPYD and DPD enzyme activity. 18 All patients in this study were genotyped for the four clinically relevant DPYD variants and phenotyped with a DPD enzyme-activity assay using ex vivo peripheral blood mononuclear cells (PBMCs). The present study subsequently analysed treatment and toxicity data of these (and additional) patients. Patients with incomplete results of genotyping and phenotyping were excluded.
Dose adjustments were made based on genotype and phenotype results by the treating physicians according to national guidelines. 25 In our analysis, the dose recommendations from the DPWG guidelines valid at the time of treatment were used. In some patients, due to clinical urgency, treatment had to be started right away without the genotyping and phenotyping results available; these patients were given a “safe start” (treatment with a lower dose, awaiting results of genotyping and phenotyping).
DPYD gene variant and DPD enzyme activity assays
Both the DPYD gene analysis and enzyme activity test were performed in one academic hospital. DNA was isolated according to standard methods and DNA of the patients was analyzed using Sanger sequencing for the four clinically relevant DPYD variants: DPYD*2A (NM_000110.4(DPYD):c.1905+1G>A), DPYD*13 (NM_000110.4(DPYD):c.1679T>G), c. 2846A>T (NM_000110.4), and c.1129-5923C>G (NM_000110.4). An extensive description of the genotyping methods was previously published. 18 In our study *1/*1 refers to patients who are not carriers of any of the four tested variants.
The DPD enzyme-activity assay using ex vivo PBMCs was performed with isolated lymphocytes. Patients were classified as having a decreased DPD enzyme activity when the enzyme activity was below 8.69 nmol/mg protein/h (<70% of the mean DPD activity in our reference cohort, which is 12.4 nmol/mg protein/h). The reference cohort and phenotyping methods are described in a previous publication. 18
Toxicity grading
The toxicity was graded on a 1–5 scale according to the CTCAE criteria, version 5.0. 4 All adverse events that were possibly, probably, or definitely related to the fluoropyrimidine treatment, as described by the physician or qualified nurse practitioner, were registered. Severe toxicity refers to CTCAE grade 3 or higher. All ≥ grade 3 events were included, clinical as well as laboratory abnormalities. Radiotherapy was mentioned as concomitant treatment when this was applied during fluoropyrimidine treatment or up to two weeks before treatment.
Patient groups
Based on the presence of DPYD variants and DPD enzyme activity measured in PBMCs, the patient cohort was divided into four groups. The first group was the control group, consisting of patients who carry none of the four tested DPYD variants and in whom a normal DPD activity was measured in PBMCs (DPYDvariant_no-DPDnormal_activity). The patients of the second group carried one of the four clinically relevant DPYD variants and had a normal DPD activity in PBMCs (DPYDvariant_yes
The total group of patients carrying a variant (regardless of DPD activity) was designated DPYDvariant_yes, the total group of patients with decreased DPD activity (with or without a DPYD variant) was designated DPDlow_activity.
Results
Relevant cohort characteristics
A total of 228 patients treated with fluoropyrimidines were included in this study, consisting of 182 patients in the DPYDvariant_no-DPDnormal_activity group, 16 in the DPYDvariant_yes
Patient characteristics.
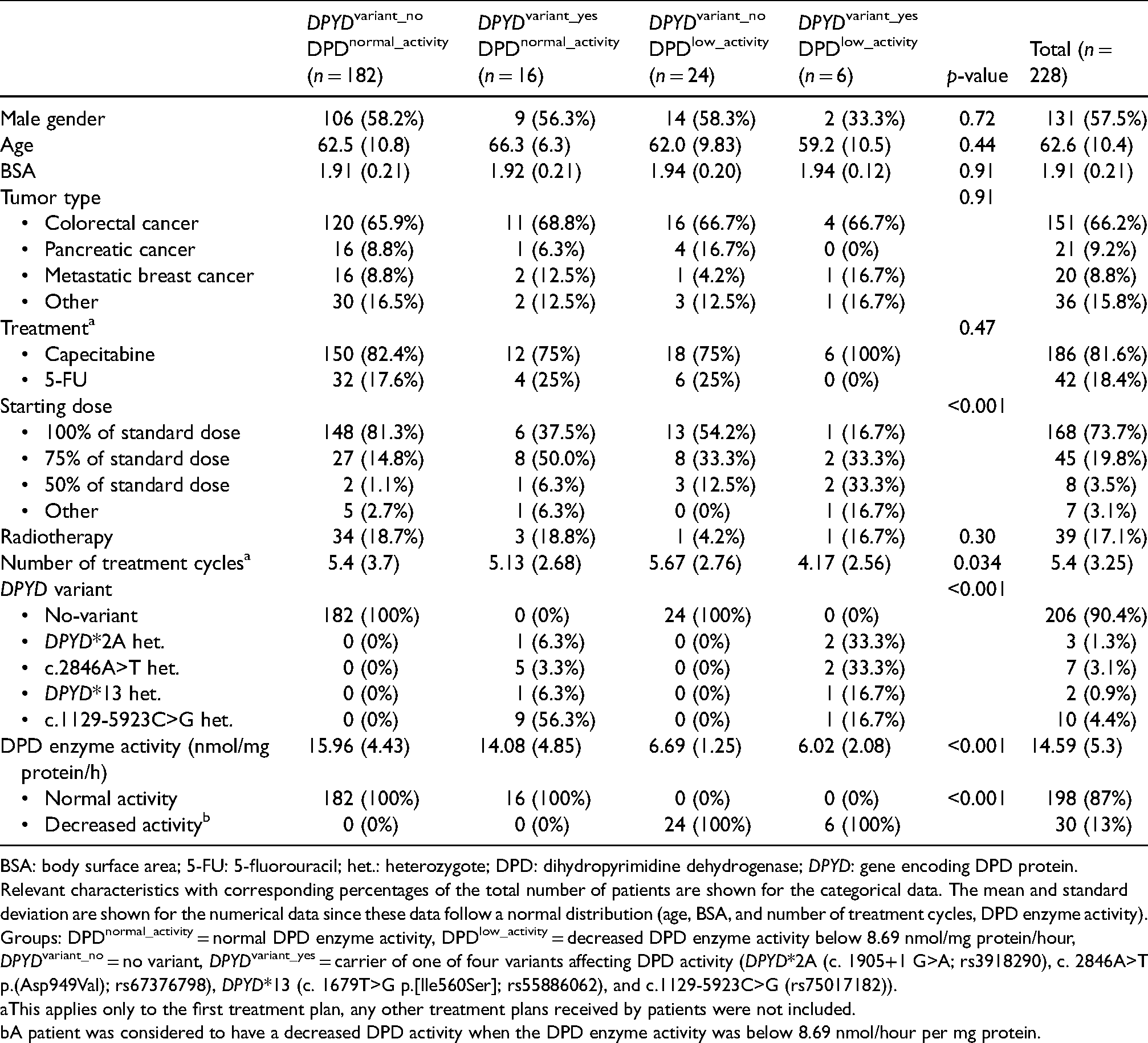
BSA: body surface area; 5-FU: 5-fluorouracil; het.: heterozygote; DPD: dihydropyrimidine dehydrogenase; DPYD: gene encoding DPD protein.
Relevant characteristics with corresponding percentages of the total number of patients are shown for the categorical data. The mean and standard deviation are shown for the numerical data since these data follow a normal distribution (age, BSA, and number of treatment cycles, DPD enzyme activity). Groups: DPDnormal_activity = normal DPD enzyme activity, DPDlow_activity = decreased DPD enzyme activity below 8.69 nmol/mg protein/hour, DPYDvariant_no = no variant, DPYDvariant_yes = carrier of one of four variants affecting DPD activity (DPYD*2A (c. 1905+1 G>A; rs3918290), c. 2846A>T p.(Asp949Val); rs67376798), DPYD*13 (c. 1679T>G p.[Ile560Ser]; rs55886062), and c.1129-5923C>G (rs75017182)).
This applies only to the first treatment plan, any other treatment plans received by patients were not included.
A patient was considered to have a decreased DPD activity when the DPD enzyme activity was below 8.69 nmol/hour per mg protein.
Colorectal cancer was the most frequent cancer reported in 66% of the patients, followed by pancreatic and breast cancer. The vast majority of the patients received treatment with capecitabine (82%), while a smaller proportion was treated with intravenous 5-FU. Standard initial treatment dose was received by 74% of the patients, while 26% of the patients received an initial dose reduction, median 75% (range 25–84%). The average body surface area (BSA) was 1.9 m2, which is slightly higher than that of the average Dutch adult population (1.7 m2). 30
DPYD variant carriers and decreased DPD enzyme activity measured in PBMCs
Twenty-two patients carried one of the four DPYD gene variants associated with decreased DPD activity and 32 patients had a decreased DPD activity measured in PBMCs. No homozygous DPYD variant carriers were identified in our cohort, nor patients with zero DPD enzyme activity. 6/22 heterozygous DPYD variant carriers (27%) showed a decreased DPD enzyme activity measured in PBMCs (DPD activity was below 8.69 nmol/per mg protein/hour) (Figure 1). More specifically, 29% of the c.2846A>T variant carriers and 10% of the c.1129-5923C>G variant carriers showed a decreased DPD enzyme activity. Of the DPYD*2A and DPYD*13 variant carriers, 67% and 50% of patients had a decreased DPD enzyme activity, respectively. There was a broad range of DPD activity in non-variant carriers.
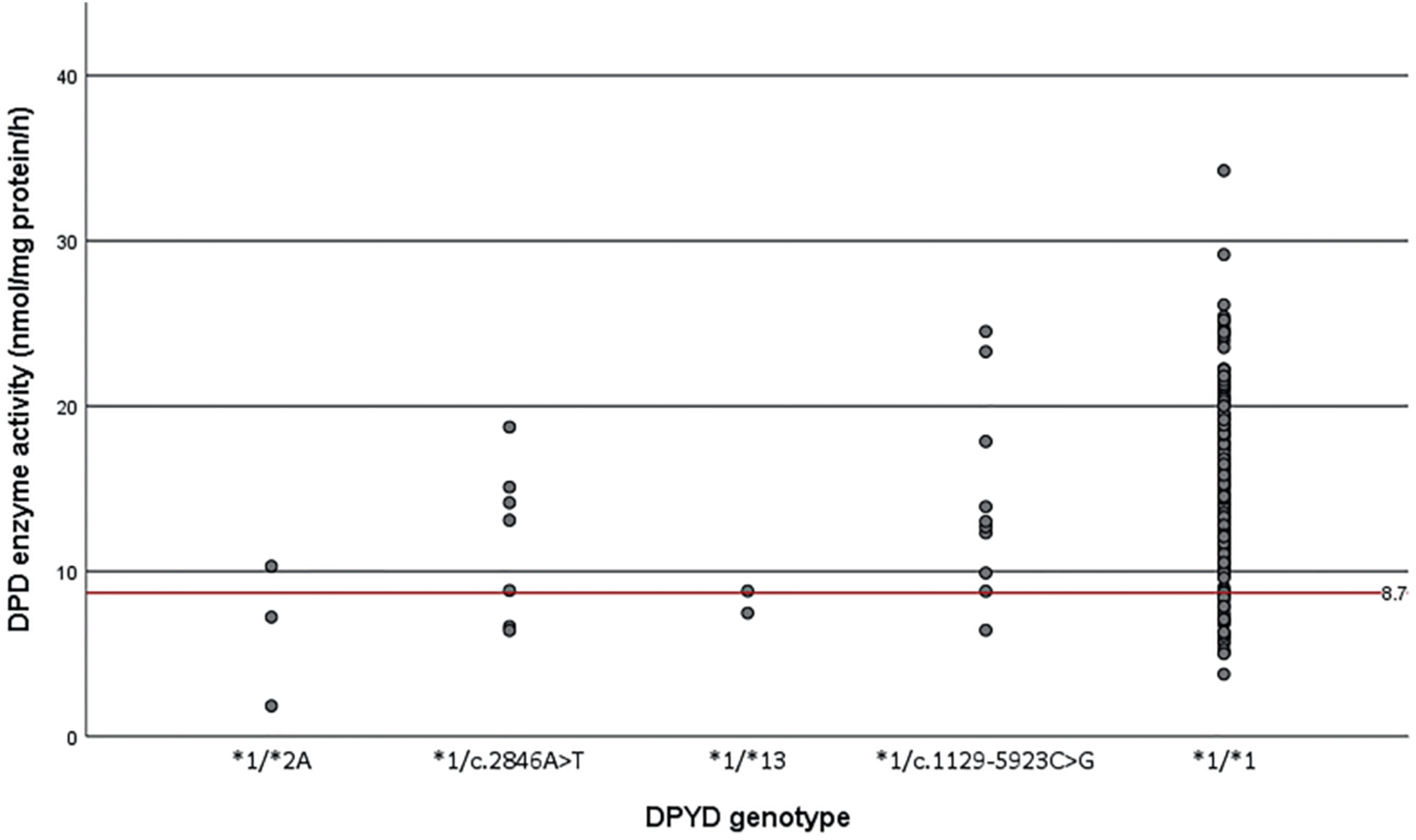
Results of the DPD enzyme activity assay using ex vivo PBMCs are shown on the y-axis, the different DPYD genotypes that are known to influence DPD enzyme activity can be seen on the x-axis: DPYD*2A/*1 (c. 1905+1 G>A; rs3918290), c. 2846A>T/*1 p.(Asp949Val); rs67376798), DPYD*13/*1 (c. 1679T>G p.[Ile560Ser]; rs55886062), c.1129-5923C>G/*1 (rs75017182) and *1/*1. The dots represent patients who are all heterozygous carriers of one of the four variants. The red line represents the cut-off value for decreased DPD activity (<8.69 nmol/mg protein/h).
Dose reductions and toxicity in different patient groups
Dose reductions and the occurrence of severe toxicity in the different patient groups are shown in Table 2. The different types of toxicity and toxicity scores are shown in Supplemental Table 2.
Dosage and occurrence of severe toxicity of the four different patient groups.
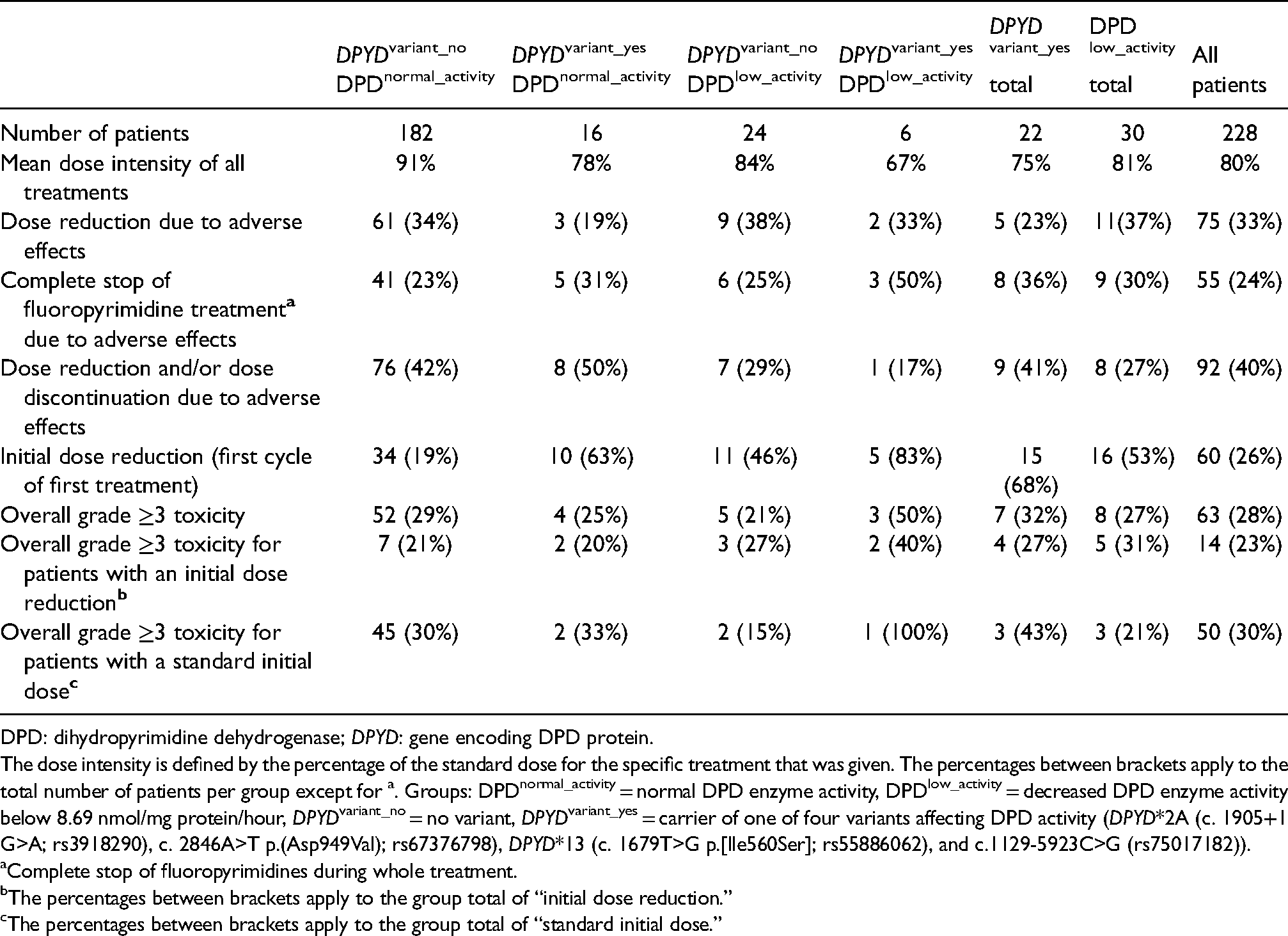
DPD: dihydropyrimidine dehydrogenase; DPYD: gene encoding DPD protein.
The dose intensity is defined by the percentage of the standard dose for the specific treatment that was given. The percentages between brackets apply to the total number of patients per group except for a. Groups: DPDnormal_activity = normal DPD enzyme activity, DPDlow_activity = decreased DPD enzyme activity below 8.69 nmol/mg protein/hour, DPYDvariant_no = no variant, DPYDvariant_yes = carrier of one of four variants affecting DPD activity (DPYD*2A (c. 1905+1 G>A; rs3918290), c. 2846A>T p.(Asp949Val); rs67376798), DPYD*13 (c. 1679T>G p.[Ile560Ser]; rs55886062), and c.1129-5923C>G (rs75017182)).
Complete stop of fluoropyrimidines during whole treatment.
The percentages between brackets apply to the group total of “initial dose reduction.”
The percentages between brackets apply to the group total of “standard initial dose.”
The mean dose intensity was highest in the DPYDvariant_no-DPDnormal_activity group (91%), followed by 84% in the DPYDvariant_no-DPDlow_activity, 78% in the DPYDvariant_yes
Overall, the percentage of patients who developed severe toxicity was 32% in DPYD variant carriers (DPYDvariant_yes), 27% in patients with decreased DPD activity (DPDlow_activity), and 29% in the DPYDvariant_no-DPDnormal_activity group. More specifically, ≥grade 3 toxicity occurred in 25% of the DPYDvariant_yes-DPDnormal_activity group and in 21% of the DPYDvariant_no
Schematic overview of the mean dose intensity and frequency of ≥grade 3 toxicity in the four different patient groups.

In the DPYDvariant_no-DPDnormal_activity group, 21% of patients with initial dose reduction experienced severe toxicity versus 30% treated with a standard initial dose. About half of the DPYDvariant_no-DPDnormal_activity patients who received an initial dose reduction (18/34) had a “safe start” (a lower dose awaiting genotyping and phenotyping results), the remaining for other reasons, for example general condition of the patient or laboratory abnormalities (Supplemental Table 1).
In the DPYDvariant_yes-DPDnormal_activity group, results were similar: 20% of patients with an initial dose reduction experienced severe toxicity, which was 33% in patients treated with a standard initial dose. In the DPYDvariant_no
Toxicity in DPYDvariant_yes-DPDlow_activity group
Fifty percent of the patients with a clinically relevant variant and a decreased enzyme activity (DPYDvariant_yes-DPDlow_activity group) experienced severe toxicity; even though they received the lowest initial dose and whole treatment dose intensity. Furthermore, 50% (3/6) of the patients in this group completely discontinued fluoropyrimidine treatment due to adverse events. This is much less frequent in the other groups, in which 23–31% of patients discontinued treatment due to adverse events (Table 2). Of the patients with a variant and decreased DPD activity, 40% (2/6) with an initial dose reduction experienced ≥grade 3 toxicity, as did the one patient who received a standard initial dose.
Of the three patients with severe toxicity in the DPYDvariant_yes-DPDlow_activity group, two experienced grade 4 toxicity and one patient died (grade 5 toxicity). In the other groups, grade 4 and 5 toxicity was very rare (Supplemental Table 2). Of note, two of these three patients from the DPYDvariant_yes
Discussion
There are very few studies investigating toxicity in patients who are genotyped and phenotyped for DPD prior to fluoropyrimidine treatment. The results of this retrospective cohort study suggest that patients carrying a DPYD variant who also have a decreased DPD enzyme activity, might have a high risk of toxicity even with a reduced fluoropyrimidine dose intensity. These patients also seem to have more grade 4–5 toxicity. Our results indicate that in patients with a known DPYD variant, the risk of severe toxicity could be even higher when the DPD enzyme activity is also low.
Our results indicate that DPD phenotyping is beneficial in identifying DPYD variant carriers with a decreased DPD enzyme activity. Formal conclusions with regards to the added value of pre-treatment phenotyping in patients without a DPYD variant in individualizing fluoropyrimidine treatment require a larger sample size. Currently, a prospective clinical trial is ongoing in the Netherlands to investigate this (clinicaltrials.gov NCT04194957). In addition, information about cost-effectiveness of additional DPD phenotying is lacking. Nevertheless, there are important advantages of phenotyping, one being able to identify patients at risk of severe toxicity who cannot be identified with standard DPYD genotyping (which was 9% of patients in a previous study 18 ). These include patients with rare DPYD variants or patients of other ethnic backgrounds. Phenotyping is also useful when a patient carries two DPYD variants associated with decreased DPD activity; in this case phenotyping is recommended by the DPWG. 25
Some drawbacks exist with regards to the utility of the DPD phenotyping method in the clinic. 19 The ex vivo PBMC assay is considered a labor-intensive method for which one needs special equipment, which is not available in every hospital, which makes it difficult to implement this method on a large scale. A cut-off value of <70% below the population mean is used to classify patients as having a decreased DPD activity, 15 which predicts fluoropyrimidine-associated toxicity,16,17 but no prospective study has been performed to determine whether this is the optimal threshold. For patients with a decreased DPD activity but no DPYD variant, there is little evidence with regards to optimal dosing strategies. The CPIC guidelines recommend a dose reduction in patients with a DPD enzyme activity <70% of normal DPD activity in the population, but no clear cut-off values are available to determine the optimal starting dose. 26 In one review several phenotype-based approaches are compared. 19 Only for DPD enzyme measurement in PBMCs a dosing strategy is established. This method calculates the starting dose in patients with decreased DPD activity using the following formula; the percentage remaining DPD activity compared to normal/average multiplied by the standard dose (% activity present × normal dose). This method, however, has not yet been validated. In summary, the clinical validity of PBMC DPD activity has been well established, but there is no clear consensus on the optimal dose adjustment strategy based on the enzyme activity.
The four clinically relevant DPYD gene variants correlate differently with phenotypic DPD enzyme activity. Although the groups are small, we found that carriers of the c.2846A>T or c.1129-5923C>G variant rarely have a decreased DPD activity measured in PBMCs (29% and 10%), while carriers of the DPYD*2A or DPYD*13 variant more frequently have a DPD activity below the threshold (67% and 50%). This is in agreement with a previous study, in which it is shown that DPYD*2A and DPYD*13 carriers have a significantly lower Dihydrouracil/Uracil ratio indicative of DPD deficiency. 23
In the total DPYDvariant_yes group, 32% (7/22) experienced severe toxicity. Only 27% of the patients who are carriers of a clinically relevant DPYD variant have a decreased DPD activity measured in PBMCs. In a larger study, also including the patients from this study, we observed the same. 18 For the patients with a variant and a normal DPD activity it might be possible to treat with a higher dose as the activity is in the normal range despite the variant. This is something that warrants additional research.
In DPYDvariant_yes and DPDlow_activity patients, the initial dose was reduced more frequently and the whole treatment dose intensity was lower, compared to the DPYDvariant_no-DPDnormal_activity patients. Despite this, the differences in overall grade ≥3 toxicity seem to be small between these groups. This could be caused by the reduced dose; if DPYDvariant_yes and DPDlow_activity patients would be treated with standard doses, more toxicity would be expected. No large difference in treatment alterations due to adverse effects in patients with and without initial dose reduction seem to be present, but the groups are too small to demonstrate significance (Supplemental Table 3).
Patients in the DPYDvariant_no-DPDlow_activity group who started treatment with the standard initial dose unexpectedly experienced less severe toxicity than DPYDvariant_no-DPDlow_activity patients with a standard initial dose. This could be due to chance as a result of the small group sizes or perhaps an indication that the threshold for low DPD activity is not optimal. The difference in severe toxicity between patients with or without initial dose reduction in the different groups cannot be explained by concomitant treatment with radio-chemotherapy (known to result in decreased risk of toxicity). Moreover, the difference cannot be explained by other factors such as whole treatment dose intensity, type of medication used, or DPD activity relative to the cut-off value (Supplemental Table 4).
Our study has several limitations. First of all, the number of patients is too small for statistical analyses. This is an observational study, and our results warrant further research in a larger, prospective study. There might be a selection bias because not all DPYDvariant_no-DPDnormal_activity patients were included; however, the included patients were randomly selected and there is no significant difference in group characteristics. Another limitation of this study is that patients with a variant or decreased DPD activity did not always receive the recommended dose reduction according to existing guidelines, because sometimes DPYD/DPD testing was performed after start of treatment. Ideally, treatment should be started after obtaining genotyping and phenotyping results, but this is not always possible due to the urgency of chemotherapeutic treatment in case of severe illness. In these cases, dose adjustments are made in the second cycle based on test results.
Nevertheless, other aspects of our study are very important. This study was performed using an existing hospital population, which reflects common practice (in one hospital). Finally, this is one of the few study populations combining genotype and phenotype data, as well as treatment and toxicity data.
In most hospitals, DPYD genotyping prior to fluoropyrimidine treatment is standard practice nowadays. DPD enzyme activity measurement is used less frequently in the Netherlands. Using a combined genotype–phenotype approach could be useful in identifying patients with a DPYD variant that have an even higher risk of severe fluoropyrimidine-induced toxicity. Furthermore, phenotyping can identify patients who do not carry one of the four DPYD variants but do have a decreased, or no DPD activity (which also includes patients who carry a rare DPYD variant not included in standard genotyping). Further studies in larger cohorts are needed to provide statistical substantiation and to investigate which starting dose alterations should be considered in the DPYD variant patients with decreased DPD enzyme activity. Finally, our results underscore the importance of obtaining genotyping and phenotyping results before start of treatment, although this might not be possible in all cases.
Supplemental Material
sj-docx-1-opp-10.1177_10781552211049144 - Supplemental material for Potential added value of combined DPYD/DPD genotyping and phenotyping to prevent severe toxicity in patients with a DPYD variant and decreased dihydropyrimidine dehydrogenase enzyme activity
Supplemental material, sj-docx-1-opp-10.1177_10781552211049144 for Potential added value of combined DPYD/DPD genotyping and phenotyping to prevent severe toxicity in patients with a DPYD variant and decreased dihydropyrimidine dehydrogenase enzyme activity by Charlotte W Ockeloen, Aron Raaijmakers, Manon Hijmans-van der Vegt, Jörgen Bierau, Judith de Vos-Geelen, Annelieke ECAB Willemsen, Bianca JC van den Bosch and Marieke JH Coenen in Journal of Oncology Pharmacy Practice
Footnotes
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.