
Abstract
Standard operating procedures (SOPs) optimization for nucleic acid extraction from stored samples is of crucial importance in a biological repository, considering the large number of collected samples and their future downstream molecular and biological applications. However, the validity of molecular studies using stored specimens depends not only on the integrity of the biological samples, but also on the procedures that ensure the traceability of the same sample, certifying its uniqueness, and ensuring the identification of potential sample contaminations.
With this aim, we have developed a rapid, reliable, low-cost, and simple DNA fingerprinting tool for a routine use in quality control of biorepositories samples. The method consists of a double ALU insertion/deletion genotyping panel suitable for uniqueness, identification of sample contaminations, and gender validation. Preliminary data suggest that this easy-to-use DNA fingerprinting protocol could routinely provide assurances of DNA identity and quality in a biorepository setting.
Introduction
The development of biobanks along with the recent advances in molecular biology have enhanced the possibility to identify genetic factors for specific diseases, investigate how genes interact with environmental factors, and develop new diagnostic tests and targeted treatments (1, 2).
To achieve greater accuracy and consistency of the results, with implications on downstream genetic analysis, it is pivotal to obtain an optimal quality of the nucleic acids (DNA and RNA) extracted from human specimens. These processes require accurate quality control procedures and SOPs in the phases of collection of biological samples and in the further analytical steps (3, 4).
Since the latest decades, we have assisted to an exponential increase in the automatic and informatic system procedures in biobanks (5, 6); however, the advantages of these new technologies are still limited with respect to preventing samples misidentification. These biases may be due to: mislabeling, wrong preanalytical managing, accidental sample mixing due to manual handling, and incorrect storage (7). Technical inaccuracy may lead to a lowering of the biorepository's quality and specificity of the biological material, which may be provided to external research groups or exchanged with other biorepositories.
Therefore, the traceability and the individuation of a potential contamination of the biological samples by a biobank is a critical point for the assessment of association studies such as Genome-Wide Association Studies (GWAS) or high-throughput sequencing (8, 9). The use of genetic forensic techniques, based on statistical evaluation of polymorphic loci of human DNA, has been previously proposed to address this issue (10, 11). However, the applicability of these methods in biobanks is often limited not only due to the lack of equipment and softwares necessary for such kind of tests, but also to the cost of the specific reagents, and the high specialistic approach necessary for these analyses (11).
In the present study, by using the infrastructure of the Interinstitutional Multidisciplinary BioBank (BioBIM), we aimed at developing a reliable, simple, and not expensive DNA fingerprinting method to verify the authenticity and purity (with respect to the contamination with allogenic genetic material) of a DNA sample belonging to a biorepository. This method was created with the final aim of performing a reliable and valuable quality control in biorepositories.
Materials and Methods
Samples and DNA extraction
Blood samples were obtained from healthy donors (n=100) enrolled by the BioBIM, Department of Advanced Biotechnologies and Bioimaging, Institute of Care and Scientific Research San Raffaele, Pisana, Rome, Italy. All procedures and observations were recorded according to the ISO-9001 standards and approved by the ethical committee of the IRCCS San Raffaele Pisana. For DNA extraction an automated protocol was performed using MagNA Pure LC instrument with MagNA Pure LC total DNA isolation kit I (Roche Diagnostics, GmbH) according to the manufacturers' instructions. Concentration and quality of DNA were assessed using an ND 1000 NanoDrop spectrophotometer (Celbio). The amount of total DNA was quantified by spectrophotometric optical density (OD) at a wavelength of 260 nm.
ALU and gender-specific markers selection
Autosomal ALU insertion/deletion (I/D) polymorphisms candidates for this study were chosen on the basis of literature databases and nucleic acid sequence data available on websites:
NCBI PubMed (http://www.ncbi.nlm.nih.gov/pubmed),
NCBI dbSNP(http://www.ncbi.nlm.nih.gov/snp),
NCBI OMIM (http://www.ncbi.nlm.nih.gov/omim),
NCBI Nucleotide (http://www.ncbi.nlm.nih.gov/nuccore),
ALFRED (http://alfred.med.yale.edu/alfred/index.asp) and Ensembl (http://www.ensembl.org/index.html).
Eight ALU I/D were then selected according to their chromosomal localizations and/or on different arms of the same chromosome. Markers with an average heterozygosity ≥0.30 in different ethnic groups were also selected. The gender-typing loci DXZ4 and SRY were included in the study based on our previous results (12, 13) (Tab. I).
ALU and Gender-Specific Molecular Markers' Features Selected for the Present Study
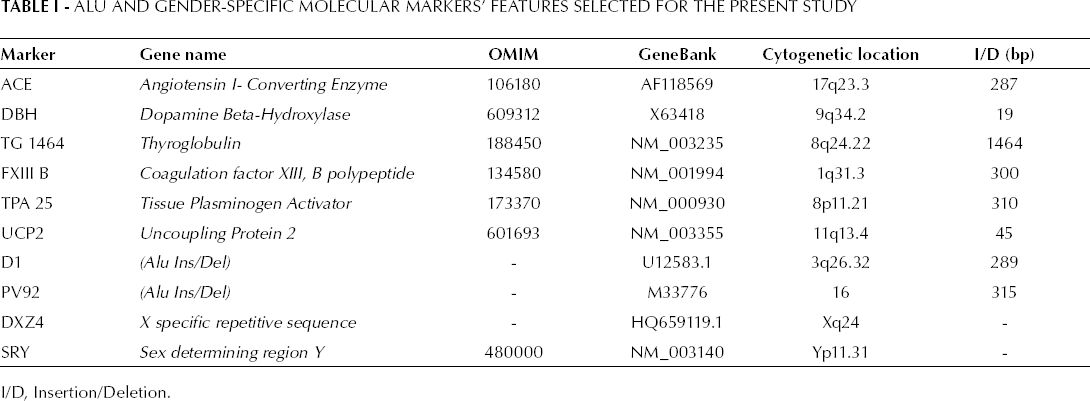
I/D, Insertion/Deletion.
Primers characteristics
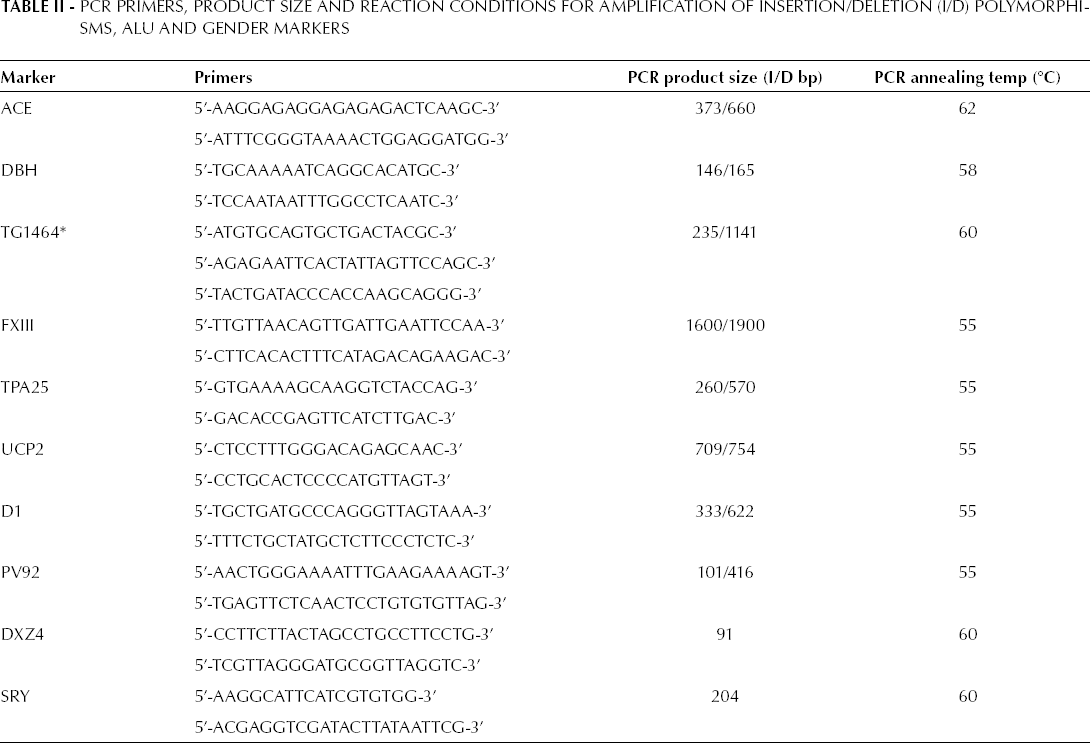
The characteristics of the primer pairs, designed for amplification of all 8 ALU sequences and the X and Y specific loci are listed in Table II. They have been classified according to their chromosomal locations, product sizes, and PCR primer sequences. All primers were designed using the MacVector 10.6.0 software (Mac Vector Inc., Cary, North Carolina, USA). The primer sequences were checked by the GenBank BLAST database (http://blast.ncbi.nlm.nih.gov/) to ensure the specificity for the respective loci.
PCR Primers, Product Size and Reaction Conditions for Amplification of Insertion/Deletion (I/D) Polymorphisms, ALU and Gender Markers
Single and multiplex PCR amplification
All the 100 selected DNA samples were typed in singleplex for each primer pair in order to evaluate primer performance, expected allele sizes and allele frequencies. PCR reactions were performed in a GeneAmp PCR System 9700 (Applied Biosystems, Carlsbad, CA, USA) using Hot-StarTaq Master Mix (QIAGEN Inc., Chatsworth, CA, USA) as follows: 32 cycles at 95°C for 30 seconds, an appropriate and primer-specific annealing temperature (Tab. II) for 30 seconds, and at 72°C for 60 seconds, with an initial denaturing step at 95°C for 15 minutes, and a final extension step at 72°C for 10 minutes.
After these evaluation assays, primer pairs were combined together and tested in multiplex. We performed various experimental tests, including different primer combinations, primer mix concentrations and annealing temperatures. Final optimizations of the assay were performed using 2 different multiplex reactions, as to avoid molecular size overload.
At the end, the final assay design resulted in 2 independent PCR-based tests, employing distinct sets of primers: Multiplex A (ACE, DBH, TG 1464, DXZ4, and SRY) and Multiplex B (FXIII B, TPA 25, UCP2, D1, and PV92).
The PCR reactions were performed in 50 μL of a mixture containing 50 ng of DNA, 25 μL of Multiplex Master Mix (QIAGEN Inc., Chatsworth, CA, USA), 5 μL of Q-solution (QIAGEN Inc., Chatsworth, CA, USA), a variable amount of distilled water and a primer mixture containing each oligonucleotide primer at a final concentration of 0.2 μM, with the exception of PV92 and FXIII B, which were used at a final concentration of 0.4 μM in Multiplex B.
The PCR reaction proceeded for 30 cycles comprising 30 seconds of denaturation at 94°C, 90 seconds of annealing at 58°C for multiplex A and at 55°C for multiplex B, 90 seconds of final extension step at 72°C for multiplex A and 150 seconds for multiplex B. An initial denaturation step of 15 minutes at 95°C and an extension step of 10 minutes at 72°C were employed. In order to exclude pre-analytical and analytical errors, all analyses were repeated on PCR products obtained from new nucleic acid extractions.
Gel electrophoresis
Amplified PCR products were separated by electrophoresis on a 2% (singleplex and Multiplex B) or 2.5% (Multiplex A) agarose gel containing ethidium bromide, and were observed by ultraviolet illumination using Quantity One 4.6.0 software and the Gel Doc XR Systems (BioRad). As size marker a Trackit™ 100 bp DNA ladder (Invitrogen) was included.
Statistical Analysis
The gene counting method was used to calculate the allele frequencies for each ALU I/D locus. The observed numbers of genotypes were compared with those expected for a population in Hardy-Weinberg equilibrium using the WEB-based tool Exact Hardy-Weinberg (https://pharmgat.org/Tools).
Statistical parameters to evaluate the Discrimination Power (DP) and Random Match Probabilities (RMPs) for each locus and multiplex profiles were calculated with a custom-made MS Excel spreadsheet (Microsoft Co., Redmond, WA, USA).
Results
By using 100 samples from healthy donors taken from BioBIM, in this study we have developed a simple, reliable, reproducible, and not expensive DNA fingerprint method that may be useful for increasing sample quality control in biobanks. The methodologies required for its application include DNA extraction, PCR amplification, and electrophoretic separation on agarose gel. In the presence of heterozygous samples, in some cases we observed the formation of mismatched heteroduplexes between the PCR products due to DNA containing insertion and deletion. All DNA profiles previously tested with singleplex PCRs were subsequently confirmed by combination of the 2 multiplex-PCRs protocol (Fig. 1). To test the reproducibility of the employed method, the DNA from the same samples was analyzed twice. Our results demonstrated that the method used is able to match individuals with a full concordance. Furthermore, gender was identified by inclusion in the analysis of the DXZ4 and SRY genes, respectively on chromosomes X and Y (Fig. 1). Based on the frequencies observed in our study population (Tab. III), each autosomic I/D markers used in this work did not significantly deviate from the Hardy-Weinberg equilibrium.
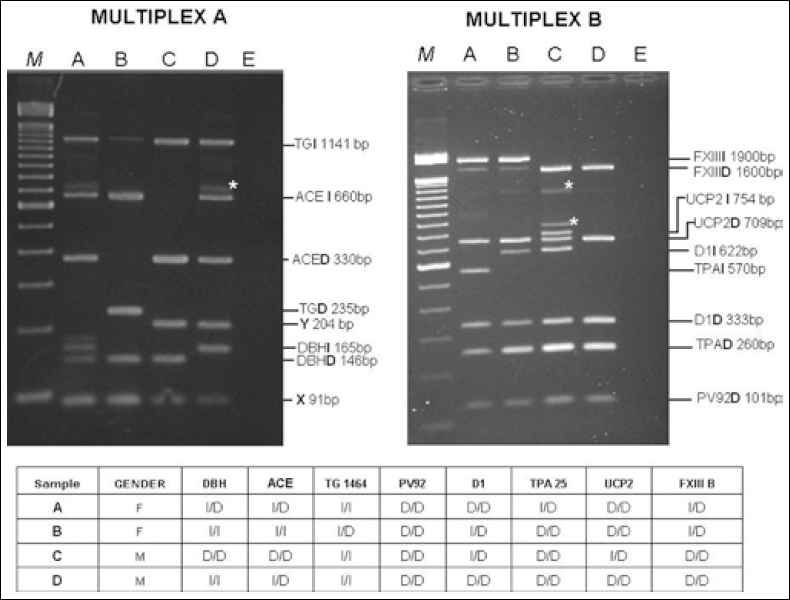
ALU insertion/deletion fingerprint and gender determination of 4 BioBim samples by multiplex PCRs. Lane M contains the Trackit™ 100 bp DNA ladder (Invitrogen); lane A-D illustrate the multiplex amplification products of samples from healthy donors taken from BioBIM; lane E contains PCR reagents and primers without DNA (negative control). ∗heteroduplex PCR bands.
ALU Id Genotype and Allele Frequencies Observed in 100 Healthy Controls from Interinstitutional Multidi-Sciplinary Biobank (BioBIM)
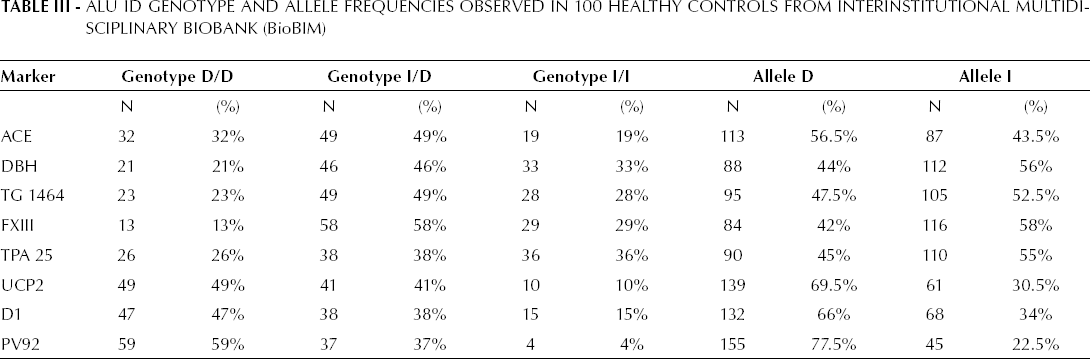
The calculated efficiency for the Multiplex A panel showed a RMP of 5.38x10−2 (1 in 20 subjects) and a DP of 94.62%. For the multiplex B we found a RMP of 1.18x10−2 (1 in 100) with a DP of 99%. The combined panels used in this study showed a cumulative RMP of 6.62x10−4 (1 in 1428) and a DP of 99.94%. By adding the gender markers to the analysis, the efficiency reached a RMP of 3.02x10−4 (1 on 3308) and a DP of 99.97%, thus providing satisfactory levels of informativeness for the requirements of a relatively limited number of samples afferent to a biobank.
To further explore the validity of the employed method we compared the genotyping results obtained from samples analyzed in 2012 with the genotyping results of samples collected in 1997 and stored at -80°C. Despite the 7 years in storage, no genotype mismatch or DNA degradation were found.
Finally, to evaluate the sensitivity of the assay in detecting contaminations, we used a group of samples specifically contaminated by combining 2 different blood samples with a serial dilution of 1:1, 1:5, 1:10, 1:25, and 1:100. The employed method was able to identify the presence of a genetic profile resulting as a combination of 2 distinct genotypes until a 1:25 dilution.
Discussion
Molecular studies, such as GWAS, necessary to identify the impact of genetic variations in large cohorts, required collaboration between different biorepositories (14). Therefore, the quality of the biological samples is pivotal to well-define the genotype-phenotype correlation in studying characteristic diseases. With this final goal, certificated procedures should be used to ensure the uniqueness of samples and to avoid the presence of contamination between different samples (15). To date current technologies are not fully applied to address this issue. In our study we tested an efficient methodological approach to identify DNA fingerprint. This method has been shown to be easy to use, not expensive, reliable, and highly reproducible.
Despite the large amount of studies performed on DNA genotyping techniques (10) and the large number of commercial systems for DNA genotyping, to our knowledge up to now there are only few studies evaluating DNA profiling in a Biobank context, and most of them are centered on cell line authentication (11, 16, 17). As an example, in a published study DNA short tandem repeats (STR) along with the gender determination (Amelogenin) gene were used to establish a reproducible approach for the authentication of human cell lines without focusing on other biobanking human samples (16). Cardoso et al (11) aimed to test a method to authenticate blood samples in biobanking processes. The authors of this study demonstrated how blood spots stored in 3 different support media did not show notable degradation signs, irrespective of the material used, suggesting that new methods can be applied in maintenance of biological sample for biobanking networks. However, the main issue of this genetic profiling system, performed by using commercial kits routinely employed in forensic medicine, arises when it becomes necessary to process a large amount of samples, as this is an expensive, and labor-intensive technique. Using a combination of ALU polymorphisms Mathot et al, in an interesting research (17), developed a robust multiplex technique, similar to the methodology employed in the present study for the detection of I/D polymorphisms, aimed at avoiding the discrepancy between normal and tumoral tissues by targeting STR in oncological subjects with microsatellite instability. Nevertheless, to fully perform such kind of investigation, a sample handling robotic platform, capillary electrophoresis automated sequencing, and high qualified personnel was required, making its spreading difficult for most of the traditional biobanks (17).
On the contrary, our method is not complicated and is usable by regular personnel in common infrastructures, as it uses techniques that are regularly present in any laboratory (DNA extraction, PCR amplification and separation by agarose gel electrophoresis), without the requirement of additional equipment or softwares. These requirements are in line with the actual economic status, in which the majority of biorepositories must operate within a limited budget in a high-priced field of research still technologically innovative and constantly expanding (18). Therefore, in many cases it seems impossible to bear the costs for performing an extremely expensive and laborious genetic profile of biosamples (11). Finally, this particular field of biomedicine inevitably requires specialized genetic expertise (which are not always present) for the production and interpretation of reliable and reproducible results.
The strength of our study includes the possibility to enhance intra-laboratory quality controls in each biobank by implementing SOPs associated to traceability of samples. In addition, the method provides the possibility of carrying out inter-laboratory quality controls as a valuable tool for comparing samples of uncertain origin or samples mismatched during previous exchanges between different biobanks or research groups. Moreover, the use of simple and informative small ALU I/D loci are analyzable through a simple PCR amplification and electrophoresis, without the need of using a sequencing equipment (19). DNA analysis of a significant number of samples could be performed spending less than 1 working day and at a cost of about US $3 per sample analyzed, an amount significantly lower than those reported in other, similar studies (7).
We also need to acknowledge some limitations of the present study. When all I/D polymorphism and X/Y chromosome markers of our study were tested the average random match probability was lower than that reached by multiplex commercial assays. This implicates that this method cannot be used for forensics purposes. However, the RMP and DP obtained by our multiplex assay provide satisfactory levels of informativeness for biobank requirements.
When heterozygous samples were analyzed by electrophoretic assay, sometimes we observed the presence of additional PCR products of variable intensity for the ALU markers UCP2, DBH, FXIII, and D1. These heteroduplex products, determined by the annealing of single-stranded I/D DNA molecules during PCR amplification, show an electrophoretic mobility that is lower than those of homoduplexes formed from the I/I or D/D homozygous samples (20). However, the detection of an additional band does not affect the interpretation of the results, since it does not overlap with any of the specific PCR products (Fig. 1).
Despite the high levels of automation currently achieved by biobanks, we decided to adopt a manual method, considering its low cost and satisfactory efficiency. Moreover, in order to further limit the costs, the method could not necessarily be applied indiscriminately to all biological samples but only to those with issues about traceability.
To our knowledge, this is the first report where ALU loci, simply detected by PCR and subsequent gel electrophoresis separation, are successfully employed for biopreservation purposes. Further studies are needed to corroborate and implement the results of this pilot study and to further verify the applicability of this method in biorepositories quality control.
Footnotes
Acknowledgments
We wish to thank Francesco De Angelis for his excellent technical assistance.