
Abstract
Familial adenomatous polyposis (FAP) is an autosomal dominant disorder related to germline mutations of the adenomatous polyposis coli (APC) gene. It is characterized by the detection of numerous adenomatous polyps that, if untreated, develop into colorectal cancer. We studied an Italian family with FAP history and the related colorectal tumor sample of the proband. Sequencing analysis of blood samples revealed the presence of a never-reported germline mutation in the APC gene (exon 15): an heterozygous G deletion at position c.2126 resulting in a premature stop codon (p.Gly721GlufsX6) and in a truncated protein. This mutation was also identified in the colorectal tumor tissue, together with a second known pathogenic heterozygotic somatic mutation, c.4348C>T (p.Arg1450X), which generates a premature truncated protein. The novel identified germline mutation is therefore related to FAP and, in accordance with Knudson's “two hit” hypothesis, can be considered the first event predisposing to the insurgence of colorectal cancer in these patients. The somatic hit inactivating the second allele of the APC gene is located in the mutation cluster region of the gene; this is not a random event since it depends on the position of the germline mutation. The inactivation of APC generates the neoplastic growth advantage to the cell.
Introduction
Familial adenomatous polyposis (FAP) is an autosomal dominant disorder characterized by the detection of hundreds to thousands of adenomatous polyps in the colon and rectum of affected individuals. Untreated, colorectal cancer invariably develops, in general in the fourth decade of life, as a consequence of additional events related to the original germline mutation. The neoplastic risk is usually related to the polyps number (1).
The genetic basis of FAP has been associated with germline mutations of the adenomatous polyposis coli (APC) gene (OMIM 175100), which is a tumor suppressor gene located on the long arm of chromosome 5 in band q21. The coding region covers 8,535 nucleotides, spanning 15 exons for a protein of 2,843 amino acids with a molecular weight of 310 kDa. Exon 15 encompasses more than 75% of the coding sequence and is the most common target of mutations (2). The APC protein has different cellular functions, it plays a central role in Wnt signaling, in part by regulating the degradation of β-catenin and, as a consequence, the transcription of a number of important cell-proliferation genes. It plays a role also in cell-cell adhesion, stability of the microtubular cytoskeleton, in addition to cell cycle regulation and possibly apoptosis (3).
Materials and Methods
We studied an Italian family after having obtained informed consent from all analyzed members (Fig. 1). The proband, patient II-1, was affected by colorectal cancer and underwent surgery for subtotal colectomy. Anatomopathological analysis indicated the presence of an ulcerated, poorly differentiated adenocarcinoma of the large intestine (pT4N0). The cancer infiltrated the intestinal wall until the visceral peritoneum. Multiple tubulovillous adenomas with a moderate dysplasia were also revealed. The tumor sample showed also the presence of codon 12 KRAS mutation GGT>TGT (G12C) detected by direct sequencing. Patients III-2 and III-4, the proband's sons, underwent colonoscopy that showed numerous polyps from the rectum to the cecum; histological analysis confirmed the adenomatous aspect of the polyps, consistently with FAP. In particular, this case can be considered as atypical FAP, according to what reported by Friedl and Aretz in 2005 (4), since it did not meet the criteria for either typical (>100 polyps and age <35), or attenuated FAP (10-100 polyps and age <25, or >100 polyps and age >45). In fact, patients III-2 and III-4 showed a large number of polyps (>100) diagnosticated at an age between 35 and 45 years, which could fit the criteria defining an atypical FAP (4).
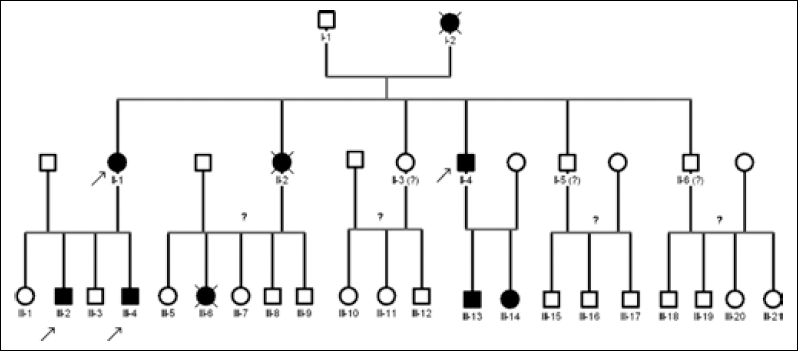
Pedigree of the studied FAP family. Each patient is indicated with the generation number followed by a progressive numeration. Solid symbols indicate patients with colorectal polyposis, arrows indicate screened patients included in this study. Individuals without information about the disease are indicated by question marks.
In the same family patients I-2, II-2, and II-4 were affected by FAP-related colorectal cancer, and I-2 and II-2 died because of the tumor; also patients III-6, III-13, and III-14 were affected by FAP (however, it is not known whether III-6 died because of colon cancer). No information has been reported about patients II-3, II-5, II-6 and their progeny.
Genomic DNA from proband II-1 and patients II-4, III-2, and III-4 was isolated from peripheral blood leukocytes using a commercial DNA isolation Kit (Roche, Italy). The proband's formalin fixed and paraffin embedded histological sections of colorectal cancer were obtained for DNA extraction (Roche, Italy), the tumor area was selected and manually microdissected by the pathologist. Mutation analysis of exon 1-15 of the APC gene (RefSeq NM_000038.4) was performed using direct sequencing technology on DNA amplification products obtained by polymerase chain reaction (PCR) from tissue and leukocytes' samples. In 20 μL PCR final reaction we used AmpliTaq Gold PCR Master Mix in a final concentration of 1X (Applied Biosystem, USA), glycerol 5%, 5 pmol of each primer, 40-80 ng of extracted DNA and water to reach the final volume. Primers were designed to include each exon flanked by a small portion of the corresponding introns, while the last large exon 15 was divided into 23 overlapping fragments. Reactions were carried out in a thermal cycler (Applied Biosystem, USA) as follows: 1 denaturing step at 95°C for 10 minutes, 5 cycles of the denaturing step at 95°C for 30 seconds, annealing step at 59°C for 45 seconds and extension step at 72°C for 45 seconds; then 30 cycles of the denaturing step at 95°C for 30 seconds, annealing step at 55°C for 45 seconds, extension step at 72°C for 45 seconds and the last 5 cycles of denaturing step at 95°C for 30 seconds, then annealing step at 53°C for 45 seconds and extension step at 72°C for 45 seconds with the final extension step at 72°C for 10 minutes. Automated cycle sequencing for both DNA strands was performed with ABI Prism® 310 Genetic Analyzer (Applied Biosystem, USA) using the Big-Dye® Terminator v1.1 Cycle Sequencing Kit (Applied Biosystem, USA) in accordance with the manufacturer's instructions. Sequence analyses were performed by Sequencing Analysis software v5.1 and Seq Scape software v2.5 (Applied Biosystem, USA). The mutation was identified as new by referring to the LOVD-IARC database (http://chromium.liacs.nl/LOVD2/colon_cancer/) and to the UMD-APC mutations database (http://www.umd.be/APC/). The mutation found was confirmed by repeated analysis in a second independent experiment of a second blood sample.
Results
Sequencing analysis revealed the presence of the following mutations: first, several non pathogenic (synonymous) known mutations, c.1458C>T (p.Tyr486 in exon 11), c.1635A>G (p.Ala545 in exon 13), c.5034A>G (p.Gly1678 in exon 15), c.5267G>T (p.Ser1756 in exon 15), c.5880A>G (p.Pro1960 in exon 15) and, second, a known mutation with uncertain pathogenic effect, c.5465A>T (D1822V in exon 15). All these mutations were found in homozygosis in proband II-1, patients III-2, and III-4, and in heterozygosis in patient II-4 (Tab. I). Moreover, a new germline mutation was found in exon 15 of the APC gene as an heterozygous G deletion at position c.2126; this mutation was found in the blood samples of all the screened patients of the family (Fig. 2A). This mutation causes a frameshift in the coding sequence, resulting in a stop codon (p.Gly721GlufsX6) and generating a premature truncated protein lacking about 75% of its sequence. This p.Gly721GlufsX6 mutation has not been so far recorded in literature and in known databases. All identified variants as well as their genotype-phenotype correlation are reported in Table I.
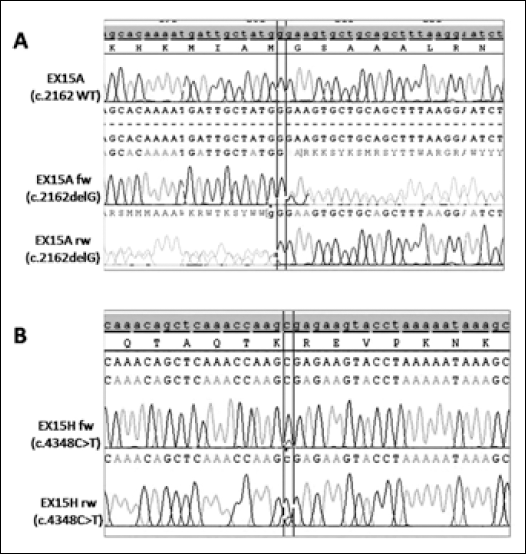
A. Novel germline APC exon 15 c.2162delG heterozygous mutation analyzed by SeqScape v.2.5 Software. The upper panel shows the reference sequence, while electropherograms show the wild type sequence (forward strand) and the mutated sequence (forward and reverse strands). B. Somatic APC exon 15 c.4348C>T heterozygous mutation analyzed by SeqScape v.2.5 Software. The upper panel shows the reference sequence, while electropherograms show the mutated sequence (forward and reverse strands).
Analyzed Patients and Genotype-Phenotype Correlation
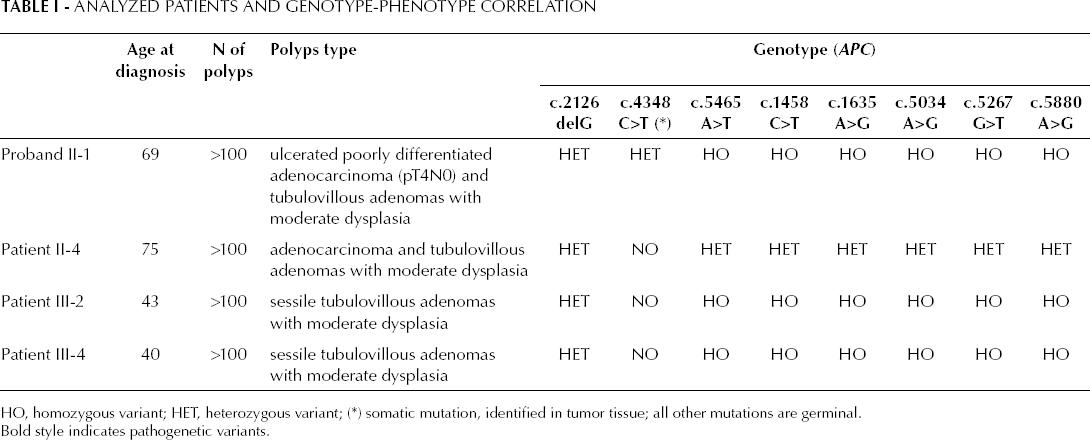
HO, homozygous variant; HET, heterozygous variant; (∗) somatic mutation, identified in tumor tissue; all other mutations are germinal. Bold style indicates pathogenetic variants.
The tumor tissue of proband II-1 showed the presence of the same germline mutations indentified in blood (both known homozygous mutations reported above and the newly identified heterozygous c.2126delG). Furthermore, a second known somatic mutation was found, in heterozygosis, in the tumor sample (Tab. I): c.4348C>T (p.Arg1450X). This variant generates a premature truncated protein and it is known as a pathogenic mutation (Fig. 2B).
Discussion
According to APC structure, the p.Gly721GlufsX6 mutation is located in the genetic sequence coding the armadillo domain of protein, consisting of 7 repeats that bind a regulatory subunit of protein phosphatase 2A (PP2A) involved in β-catenin homeostasis. In fact, in the absence of a functional APC protein, β-catenin is stabilized and accumulates in the cytoplasm and nucleus. This results in uncontrolled transcriptional activation of different genes, such as c-myc and cyclin D1, which may contribute to cell proliferation and cancer progression. It is probable that the armadillo domain plays an important role in the tumor suppressor function of APC (3).
The pathogenic role of this novel mutation is further supported by data in the literature that report similar variants causing the creation of a stop codon very close to the one observed in our case, and related to FAP (5). Moreover, the colon cancer gene database, LOVD, reports as pathogenic a c.2154delT mutation (p.Ile718MetfsX9) that is responsible for the formation of a stop codon at position 727, exactly the same one created by p.Gly721GlufsX6.
Tumor tissue molecular analysis revealed the presence of both KRAS G12C mutation and a second APC mutation. The first is a G>T transversion at nucleotide 34 of the KRAS gene, which is generally associated with MUTYH-associated-polyposis (MAP). In the case of MAP, the frequency of G12C mutation raises by increasing the degree of dysplasia and it is associated with tubulovillous morphology (6). Although this mutation is not very frequent in FAP, in the case we report here, the presence of the G12C mutation could be related to the tubulovillous morphology and moderate (even not severe) dysplasia of adenomas from which the adenocarcinoma is supposedly evolved in the proband.
Moreover, in the analyzed tumor tissue, both alleles of the APC gene were inactivated because of the presence of a second stop codon mutation, p.Arg1450X. Over 60% of all somatic mutations of the APC gene occur between codons 1286 and 1513; this region is termed mutation cluster region (MCR) (7). Within the MCR there are 2 hotspots for somatic mutations at codons 1309 and 1450 (8, 9). APC mutations within the MCR result in a truncated APC protein that lacks all of the axin binding sites and all, except 1 or 2, of its 20-aminoacid β-catenin binding sites (3). It has also been found that FAP patients with germline APC mutations within a small region (codons 1194-1392) mainly show allelic loss (LOH) in their colorectal adenomas, whereas tumors with mutations outside this region tend to harbor a truncating mutation in MCR (10). The second relationship is the one observed in our case, as the germline mutation was identified at codon 721, while the second somatic mutation was found at codon 1450. When combined with other previously reported data (11), our results show the interdependence of the “two hits” at the APC gene in colorectal tumors associated with FAP.
In conclusion, the novel familiar germline mutation p.Gly721GlufsX6, identified in the analyzed members of the family with confirmed diagnosis of intestinal polyposis, is related to FAP and, in accordance with Knudson's “two hit” hypothesis supported by the biallelic gene inactivation found in the tumor tissue, can be considered the first event predisposing to colorectal cancer in FAP patients.
Footnotes
The research work described in this manuscript was performed according to approved scientific guidelines.