
Abstract
Aberrant DNA hypermethylation at gene promoters is a frequent event in human breast cancer. Recent genome-wide studies have identified hundreds of genes that exhibit differential methylation between breast cancer cells and normal breast tissue. Due to the tumor-specific nature of DNA hypermethylation events, their use as tumor biomarkers is usually not hampered by analytical signals from normal cells, which is a general problem for existing protein tumor markers used for clinical assessment of breast cancer. There is accumulating evidence that DNA-methylation changes in breast cancer patients occur early during tumorigenesis. This may open up for effective screening, and analysis of blood or nipple aspirate may later help in diagnosing breast cancer. As a more detailed molecular characterization of different types of breast cancer becomes available, the ability to divide patients into subgroups based on DNA biomarkers may improve prognosis. Serial monitoring of DNA-methylation markers in blood during treatment may be useful, particularly when the cancer burden is below the detection level for standard imaging techniques. Overall, aberrant DNA methylation has a great potential as a versatile biomarker tool for screening, diagnosis, prognosis and monitoring of breast cancer. Standardization of methods and biomarker panels will be required to fully exploit this clinical potential.
Introduction
Breast cancer is the most common malignancy among females in Western societies and is responsible for nearly half a million deaths each year worldwide. Recent advances in gene profiling and integration of different technology platforms have provided important insights into the molecular mechanisms of tumor development (1). Mutations that drive tumor development may be either inherited via the germline or acquired in somatic cells later in life. Only 5%-10% of all breast cancer cases have a clear inherited component, including germline mutations in the breast cancer 1 and 2 genes (BRCA1 and BRCA2) and several less well defined (“non-BRCA”) genes (2, 3). The remaining group of breast cancers (90%-95%) consists of sporadic cases that are caused by the acquisition of genetic and epigenetic changes in somatic cells.
Epigenetic changes include histone modifications (acetylation, methylation, phosphorylation, sumoylation and ubiquitylation) and changes in methylation of DNA (4, 5). DNA methylation is a covalent addition of a methyl group to a cytosine located adjacent to a guanosine in the same strand (a CpG dinucleotide). Thus, cytosine can be either methylated or non-methylated at the CpG dinucleotide motif. This motif is found throughout the genome, although with a higher density (so called “CpG islands”) at gene promoter regions. Hence, changes in CpG methylation can occur globally in the genome or locally at promoter regions. In general, breast cancer is associated with global hypomethylation and local promoter hypermethylation (4, 5). Therefore, breast cancer cells can be distinguished from normal cells by their different molecular fingerprints defined by the patterns of promoter hypermethylation events (4, 5). These unique “breast cancer” fingerprints are potentially reversible and may cause plasticity in the expression of oncogenes and tumor suppressor genes.
Many different techniques exist that allow detection and characterization of DNA methylation (6). A key method is the sodium bisulfite modification of DNA, which converts unmethylated cytosines to uracils without affecting methylated cytosines. The presence of uracil or cytosine can then be detected by methods based on restriction enzyme digestion, hybridization, sequencing or PCR (6). These techniques allow small amounts of aberrantly methylated DNA to be detected among large amounts of normally methylated DNA.
The demonstration of tumor-derived DNA in serum (7-10) and plasma (11-14) is a clinically interesting option, which may represent the basis for non-invasive detection of breast cancer. Alternatively, samples containing breast epithelial cells may be obtained non-invasively by nipple aspiration. Fine needle aspirate (15, 16), nipple aspirate (17, 18), and cells collected from lavage (19, 20) have also been investigated in the clinical assessment of breast cancer.
Thus, the sensitive methods for detection of aberrant DNA methylation in different types of samples may pave the way for clinical screening, diagnosis, prognosis and monitoring applications in breast cancer (4, 5). Here we review the most common DNA-methylation markers used in clinical studies of sporadic breast cancer.
Screening
Identification of a biomarker with a high clinical sensitivity and specificity for detection of breast cancer in an early phase provides a great potential for improving the final clinical outcome. The evaluation of DNA methylation as a screening tool is tempting since blood or oxytocin-induced nipple aspirate could be sampled from prospective cohorts. Taking into account the ability of PCR-based technologies to detect tumor-derived DNA in large amounts of normal DNA, this method could potentially be used in conjunction with an existing mammography screening program to minimize the number of false-positive cases.
Most research on DNA hypermethylation in breast cancer has focused on single copy genes located in gene promoter CpG islands. However, recent data suggest that global hypomethylation in repetitive regions may be a contributor to variation in gene expression, and repetitive elements may comprise ~45% of the human genome (21). An example of such a sequence is the long interspersed nuclear repetitive elements (LINE-1), which comprise ~10%-20% of the human genome (21, 22). Brennan et al (21) investigated LINE-1 hypomethylation in DNA from white blood cells as a marker of breast cancer risk. However, no difference in LINE-1 methylation between large cohorts of cases and controls could be observed in white blood cells, but quintile analyses for hypermethylation of the ataxia telangiectasia mutated gene (ATM) revealed an increased risk of breast cancer (21). Xu et al (22) also exploited global methylation content in LINE-1 and used peripheral blood from well-characterized cohorts; their results showed that changes in LINE-1 methylation were not associated with a higher breast cancer risk (22). However, global hypermethylation can be assessed by the luminometric methylation assay (LUMA) technique (22). Compared with women in the lowest quintile of LUMA methylation, those in the highest quintile had an increased risk of breast cancer (22). At present, further studies are required to document DNA-methylation techniques as a tool to effectively screen large cohorts of women.
Diagnosis
Diagnosis of the type of breast cancer (lobular versus ductal carcinomas), degree of invasion into the surrounding tissue, degree of metastasis, or status of the estrogen receptor (ER), progesterone receptor (PR), human epidermal growth factor receptor 2 (HER2) have been determined by measuring methylated DNA in cytological samples derived from the primary tumor, serum, blood, nipple aspirate or lavage.
As shown in Table I, many different genes have been reported to diagnose lobular or ductal carcinomas. Many of the aberrantly methylated genes play important roles in hormone signaling, cell cycle regulation, apoptosis, tissue invasion, and angiogenesis.
Ranking of the Most Common Methyl DNA Biomarkers in Clinical Breast Cancer Studies After % Sensitivity Found in the Indicated References
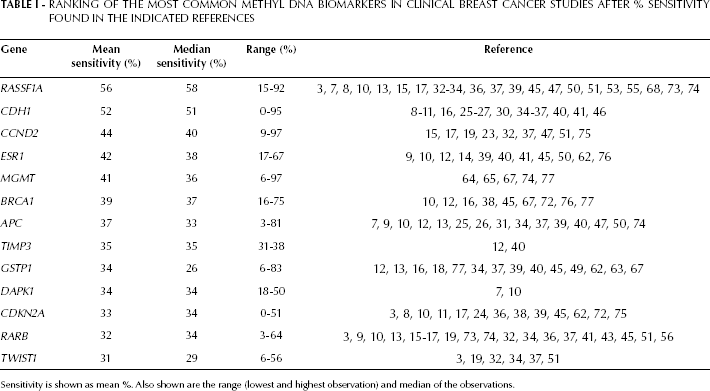
Sensitivity is shown as mean %. Also shown are the range (lowest and highest observation) and median of the observations.
One such gene encodes cyclin D2, which controls cell cycle regulation. Hypermethylation of the CpG island in the CCND2 promoter was detected by methylation-specific PCR in 46% of primary breast carcinomas and ductal carcinoma in situ. This change in methylation was associated with loss of cyclin D2 expression (23). Another gene important for cell cycle regulation is the cyclin-dependent kinase inhibitor 2A (CDKN2A), which encodes 2 tumor suppressor proteins, p16INK4A and p14ARF. Silva et al (24) examined the mRNA expression of these genes by RT-PCR in 100 primary breast carcinomas. Promoter hypermethylation was the major mechanism of inactivation of these genes and was present, respectively, in 31% and 50% of the tumors that showed decreased p16INK4A and p14ARF expression.
Downregulation of epithelial cell-cell adhesion protein E-cadherin (encoded by CDH1) may be a defining feature of lobular and ductal carcinomas (25-29). Examining microdissected archived tissue from lobular breast carcinomas and ductal tumor biopsy material by methylation-specific PCR it was found that 95% and 26% of, respectively, the lobular and ductal carcinomas had a hypermethylated CDH1 promoter (27, 28). Interestingly, CDH1 methylation was also present in non-neoplastic epithelia adjacent to methylated lesions from lobular breast cancer patients, but was absent in mammary epithelia from women without cancer (27). These results (27, 28) indicate that cell invasiveness and downregulation of the cell-adhesion capability through the E-cadherin protein are common mechanisms in breast cancer (11, 25-29). Toyooka et al (30) examined the methylation status of another cadherin family member, i.e. the heart-isoform, H-cadherin gene (CDH13). Hypermethylation of this gene was found in 33% of primary breast tumors.
Jin et al (31) investigated the methylation status of adenomatous polyposis coli gene (APC) in primary breast cancers and in their non-cancerous breast tissue counterparts. Hypermethylation was detected in 36% of primary breast cancers and in none of the 21 non-cancerous breast tissue samples. The methylation status of APC has been combined with the methylation status of CDH1 to investigate invasive lobular and ductal carcinomas (25, 26). Promoter hypermethylation of CDH1 and APC was observed in 50% and 22% of invasive ductal carcinomas, respectively (25). This panel consisting of CDH1 and APC was used in another study of lobular carcinomas, where transcription was inactivated by promoter hypermethylation in, respectively, 41% and 52% of the cases (26).
Fackler et al (32) used a panel consisting of CCND2, rat sarcoma associating domain family 1A (RASSF1A), cytokine high in normal type 1 tumor suppressor (HIN-1), retinoic acid receptor β (RARB) and twist-related protein 1 (TWIST1) to measure methylation in 2 types of lobular cancers, in situ and invasive lobular carcinomas, and compared the results with ductal in situ and invasive breast cancer. The clinical specificity was high since hypermethylated HIN-1, RARB, CCND2 and TWIST1 were not detected in any of the benign samples or mammal reduction specimens. The methylation profile of lobular vs ductal carcinomas was similar for 4 of these genes. However, TWIST1 was hypermethylated more often in invasive ductal carcinoma than in invasive lobular carcinoma. Roughly, this finding (32) suggests that hypermethylation is likely to be important in both ductal and lobular carcinomas. The highest frequency of hypermethylation in ductal and lobular in situ and invasive carcinomas was for the RASSF1A biomarker. Thus, there is a considerable interest in using the RASSF1 gene to diagnose breast cancer. RASSF1 encodes 2 major transcripts, RASSF1A and RASSF1C. Burbee et al (33) explored RASSF1A and RASSF1C in 39 resected breast cancers and found no evidence for RASSF1C as an epigenetic target. In contrast, RASSF1A hypermethylation was detected in 49% of primary breast tumors. Shinozaki et al (34) examined hypermethylation profiles of 151 primary breast tumors against RASSF1A, APC, CDH1, TWIST1, GSTP1 and RARB. The most frequently hypermethylated gene was RASSF1A (81%). RASSF1A and RARB hypermethylation was significantly associated with ER and HER2 positive tumors, whereas hypermethylation of CDH1 was significantly associated with lymphovascular invasion, ductal invasion, and lack of the ER protein. In paired sentinel lymph node metastases, CDH1 was the most frequently methylated gene (90%).
Taken together, these studies (23, 24) suggest that loss of cell cycle regulators are a part of the development of breast cancer, and that lobular and ductal carcinomas share many common methylation patterns (25-28). There is a clear correlation of CDH1 and APC promoter methylation with loss of E-cadherin and APC proteins. This points to the direct involvement of APC and CDH1 promoter methylation in early stages of breast cancer progression (11, 25-31).
Standard diagnosis of breast cancer relies on determination of the hormone receptor status. Calderio et al (35) assessed the methylation pattern of the CDH1 promoter and the standard immunohistochemical parameters of ER, HER2, and PR in 79 primary breast cancers. CDH1 hypermethylation was observed in 73% of the ductal carcinomas and in 80% of the lobular carcinomas. In the group of invasive ductal carcinomas, CDH1 hypermethylation was associated with reduced levels of expression of ER and PR (35). Feng et al (36) studied tumor-suppressor genes in 90 pairs of malignant and normal breast tissues and found 5 markers (reversion-induced LIM (RIL), HIN-1, RASSF1A, CDH13, and RARB) to be frequently hypermethylated in breast cancers, but not in normal breast tissue. Two panels of methylation profiles were defined, i.e., RIL/CDH13 and HIN-1/RASSF1A. The methylation status of HIN-1/RASSF1A strongly correlated with the expression of ERs and PRs. The subset of breast cancers negative for ER and PR correlated with the hypermethylation of RIL/CDH13. Those with negative ER, PR and HER2 status (triple-negative) were positively and negatively correlated with the hypermethylation of RIL/CDH13 and HIN-1/RASSF1A, respectively (36). Sunami et al (37) examined glutathione S-transferase (GSPT1), nestin 1 (NES1), RASSF1A, CCND2, TWIST1, APC, RARB, and CDH1. In early stages of tumor progression (T1 and N0), RASSF1A and CCND2 were significantly more frequently hypermethylated in ER-positive than in ER-negative tumors. Double ER and HER2 negative breast cancers had significantly lower frequencies of RASSF1A, GSTP1 and APC methylation (34). Jing et al (38) examined BRCA1, CDKN2A and stratifin (SNF) in the sera of sporadic breast cancer patients and healthy serum controls. The methylation frequencies of these 3 markers combined were associated with grade, according to the TNM staging system, and ER status. CDKN2A and BRCA1 hypermethylation was associated with PR status, while CDKN2A and SNF hypermethylation alone respectively correlated with histological type and lymph node metastases (38). Matuschek et al (39) prospectively obtained serum samples from patients with breast cancer as well as healthy volunteers and also examined CDKN2A together with estrogen receptor 1 gene (ESR1), APC, RASSF1A, and GSTP1. In contrast to the 4 other genes, no hypermethylation of CDKN2A was found. A significant difference was seen in methylated APC between cancer patients and healthy volunteers. Methylated GSTP1 was predominantly found in the serum of patients with large primary tumors and correlated with positive HER2 status, and the detection of methylated APC and GSTP1 in serum correlated with the presence of circulating tumor cells (39).
The degree of cancer progression and metastasis into the surrounding tissue are normally assessed by cytological examination and physical examination of the lymph nodes. This clinical information has been correlated with methylation status of various biomarkers (40-44). To assess when such methylation increases with malignant progression, Nass et al (41) surveyed 111 breast ductal carcinomas for aberrant methylation of ESR1 and CDH1. Hypermethylation of either gene was evident prior to invasion in 30% of ductal carcinoma in situ lesions and increased to nearly 60% in metastatic lesions. Coincident methylation of genes also increased significantly from approximately 20% in ductal carcinoma in situ to nearly 50% in metastatic lesions (41). Lehman et al (44) analyzed the methylation status of 4 growth regulatory genes (CDKN2A, RASSF1A, CCND2 and SNF) during breast cancer progression. When ductal and invasive tumor cells were compared, changes in methylation level were detected primarily in CCND2 (44). Widschwendter et al (43) examined RARB in 16 breast cancer biopsy specimens and non-neoplastic breast tissue. Six breast cancer specimens showed methylation in RARB and an inverse relationship between methylation and gene expression was found in all grade II lesions. No expression of RARB was found in any grade III lesion (43). Hoque et al (40) used an expanded panel of 9 genes (catenin beta 1 [CTNNB1], methylguanosine-DNA-methyltransferase [MGMT], target of methylation-induced silencing [THBS1], tissue-inhibitor of metalloprotease 3 [TIMP3], APC, CDH1, ESR1, GSTP1) and investigated changes in CpG islands promoter methylation patterns during ductal breast cancer progression. Aberrant promoter methylation was detected in both pre-invasive and invasive lesions for APC, CDH1, CTNNB1, TIMP3, ESR1, and GSTP1. CDH1 showed higher methylation levels in invasive tumors than in pre-invasive lesions. The analysis of APC and CDH1 methylation status was able to distinguish between normal and pathologic samples with a sensitivity of 67% and a specificity of 75% (45). Park et al (42) also evaluated the changes in promoter hypermethylation during breast cancer progression from pre-invasive lesions to invasive ductal carcinoma. Initial investigation consisted of analyzing the methylation status of 57 promoter CpG islands in 20 invasive ductal carcinomas and their paired normal breast tissues. Fifteen CpG islands showed breast cancer-specific changes in DNA methylation. The number of methylated genes increased stepwise from normal breast tissue to flat epithelial atypia, atypical ductal hyperplasia and ductal carcinoma in situ. The methylation status of invasive ductal carcinoma did not differ from that of ductal carcinoma in situ (42).
The findings (41, 42) showed that promoter CpG island methylation may change significantly from pre-invasive lesions towards invasion of the surrounding tissue and metastasis. Overall, the various combined panels of DNA-methylation markers (40-44) could be used to diagnose progression in breast cancer, although no clear picture emerges for the selection of the best panel of biomarkers.
Prognosis
Gene promoter hypermethylation may have additional clinical and pathological utility for assessing patient prognosis (38, 45-49). As an example, molecular alterations associated with breast cancer metastasis have been examined (38, 39, 49-52). The samplings have been based on archived breast tissues (45), nipple aspirate and lavage (47), and serum (46) to predominantly investigate the role of DNA methylation of RASSF1A (47, 53), CCND2 (47), APC (46, 47), and paired-like homeodomain transcription factor 2 (PITX2) (48, 53).
Jing et al (38) investigated the prognostic significance of BRCA1, CDKN2A and SNF in serum from sporadic breast cancer patients and healthy controls. The aberrant methylation of SNF was associated with lymph node metastases (38). Zurita et al (52) also investigated the methylation status of SNF in combination with ESR1 in metastatic breast cancer patients. The correlation of SNF and breast cancer metastasis and progression found in these 2 studies (38, 52) suggests a possible application of SNF as a biomarker to screen for metastasis and for follow-up on patients treated for metastatic breast cancer. Matuschek et al (39) prospectively analyzed APC, RASSF1A, ESR1, CDKN2A and GSTP1 and found methylated APC and RASSF1A to be differentially methylated in metastatic versus non-metastatic disease (39). Van der Auwera et al (50) investigated APC, RASSF1A, ESR1 and circulating tumor cells in blood from metastatic breast cancer patients by obtaining whole blood, plasma and serum samples. APC, RASSF1A and ESR1 were hypermethylated in, respectively, 29%, 35% and 20% of the cases. Detection of a methylated gene in serum was associated with detection of circulating tumor cells in blood (50). Mehrotra et al (51) investigated the frequency of hypermethylation of CCND2, RARB, TWIST1, RASS-F1A, and HIN-1 in metastasis to 4 common sites: lymph node, bone, brain, and lung. No hypermethylation was detected in these 5 genes in the normal host tissues. In paired samples, lymph node metastasis had a trend of higher prevalence of methylation compared with the primary breast carcinoma for all 5 genes, with significance for HIN-1 (51).
Taken together, several differentially methylated genes (APC, RASSF1A, ESR1, CDKN2A, GSTP1, CCND2, RARB, TWIST1, RASSF1A, PITX2 and HIN-1) could be validated as biomarkers for metastasis and circulating tumor cells in breast cancer patients (49-53).
DNA methylation in various promoter regions has been investigated in anti-hormonal therapies involving tamoxifen (54-59), anthracycline (60-63) and alkylating agents (64-66) to predict the efficacy of these therapies. Widschwendter et al (54) examined ESR1 methylation, which outperformed hormone receptor status as a predictor of clinical response in patients treated with the anti-estrogen tamoxifen. CYP1B1 encodes the tamoxifen and estradiol-metabolizing enzyme cytochrome P450. Promoter methylation of CYP1B1 predicted a response that differed between tamoxifen-treated and non-tamoxifen-treated patients. High levels of ADP-ribosylarginine hydrolase (ARHI) promoter methylation were strongly predictive of good survival in patients who had not received tamoxifen therapy (54). Fiegl et al (55) collected serum samples pre-therapeutically and 1 year after surgery from 148 breast cancer patients receiving adjuvant tamoxifen. RASSF1A hypermethylation was found in 19.6% of the pre-therapeutic samples and in 22.3% of 1-year-after samples. RASSF1A methylation 1 year after primary surgery (and during adjuvant tamoxifen therapy) was an independent predictor of poor outcome with a higher risk of relapse and death (55). Liggett et al (56) studied methylation in cell-free plasma DNA of 20 breast cancer patients analyzed before surgery, after surgery and after surgery on tamoxifen therapy. Tamoxifen treatment changed methylation only in the B promoter of ESR1 (56).
Martens et al (57) examined resistance to endocrine tamoxifen therapy in patients with recurrent breast cancer. Promoter hypermethylation of PSAT1 coding for a phosphoserine aminotransferase was a predictor of tamoxifen therapy response. Iorns et al (58) identified CDK10 as an important determinant of resistance to endocrine tamoxifen therapies. Maier et al (59) examined node-negative hormone-receptor-positive breast cancer patients after adjuvant endocrine tamoxifen therapy. DNA methylation of PITX2 showed the strongest correlation with distant recurrence. Its impact on patient outcome was validated in an independent cohort: 86% of patients with low PITX2 methylation were metastasis-free after 10 years, compared with 69% with high PITX2 methylation. Hartmann et al (60) investigated whether PITX2 methylation predicts outcome in node-positive, ER-positive, HER2-negative breast cancer patients who received adjuvant anthracycline-based chemotherapy. Hypermethylation of PITX2 and 14 other genes correlated with clinical outcome. Combined, these studies (59, 60) provide further evidence for PITX2 as a biomarker for both tamoxifen and anthracycline therapies.
Dietrich et al (61) found promoter hypermethylation of CDO1, encoding cysteine dioxygenase 1, to be a strong predictor of distant metastasis in retrospective cohorts of lymph node and ER-positive breast cancer patients subjected to adjuvant anthracycline-based chemotherapy. Dejeux et al (62) investigated a large panel of biomarkers on patients with locally advanced breast cancer treated with neoadjuvant doxorubicin. Absence of ATP-binding casette B1 (ABCB1) methylation correlated with progressive disease during doxorubicin treatment. Most importantly, the DNA methylation status of the promoters of forkhead box C1 (FOXC1), GSTP1, and ABCB1 correlated with survival. In multivariate analysis, GSTP1 and FOXC1 status proved to be independent prognostic markers associated with survival. Methylation of ABCB1 or GSTP1 was associated with increased overall survival, probably due to prolonged availability and activity of the drug in the cell, while FOXC1 might be a protective factor against tumor invasiveness. FOXC1 proved to be a general prognostic factor, while ABCB1 and GSTP1 might be predictive factors for the response to and efficacy of doxorubicin treatment. Arai et al (63) examined whether GSTP1 expression is associated with resistance to docetaxel or paclitaxel in 62 primary breast cancer patients. The mean tumor reduction rate for all patients was significantly higher in GSTP1-negative than GSTP1-positive tumors (63).
Fumagalli et al (64) evaluated MGMT status in 92 triple-negative breast cancer patients. MGMT encodes for a methylguanine-DNA methyltransferase that is involved in DNA repair exerted by cleavage of mutagenic alkyl adducts within DNA. It was concluded that in triple-negative breast cancer the DNA repair system may be frequently silenced by MGMT methylation (64). Esteller et al. (65) found that hypermethylation of the MGMT promoter in gliomas is a useful predictor of the responsiveness of the tumors to alkylating agents. In conclusion, MGMT hypermethylation may provide alterations in chemosensitivity to alkylating chemotherapeutic agents in many tumor types (64-66).
The results reveal a complex interaction between DNA methylation and hormone receptor biology in breast cancer cells and suggest clinically useful novel DNA-methylation predictors of response to hormonal breast cancer tamoxifen therapy (54-59), anthracycline (60-63) and alkylating agents (64-66). Pharmacoepigenetic effects, such as the reported associations in this study, provide molecular explanations for the observed differential responses to different regimens of chemotherapy, and it might prove valuable to take the methylation status of selected genes into account for treatment management. However, change in treatment management requires that alternative options of treatment are available. For example, HER2-positive patients – with a high degree of metastasis and poor prognosis – may already be treated with life-long administration of Herceptin supplemented with another chemopreventive drug.
Monitoring
Identification of biomarkers for monitoring improves measurement of the tumor burden during treatment and saves patients from toxicity from non-effective drugs. Since 80% of breast cancers are hormone-dependent, these patients are often treated with tamoxifen after surgery to target disseminated tumor cells. Disseminated tumor cells may induce a metastatic situation that will inevitably lead to death. For future secondary adjuvant treatment, a highly sensitive DNA-methylation marker for monitoring tamoxifen-resistant circulating cells could improve the survival rate. Nevertheless, only few studies have tested the clinical use of DNA-methylation markers in clinical monitoring (52, 67, 68).
Sharma et al (67) monitored methylation changes in circulating DNA in 6 consecutive serum samples collected from 30 breast cancer patients undergoing neoadjuvant chemotherapy. The patients were chosen at random in regards to ER, PR and HER2 status, and received a combination of cyclophosphamide, methotrexate, and 5-fluorouracil or a combination of cyclophosphamide, epirubicin, and 5-fluorouracil chemotherapy. Blood samples collected at the time of patient enrollment in the breast cancer clinic were considered as the baseline and were compared with blood samples collected at the end of each cycle of chemotherapy. The sera were analyzed for methylation status of a panel of 5 genes, namely BRCA1, MGMT, GSTP1, SNF and the gene coding for multidrug resistance 1 protein (MDR1) (67).
Avraham et al (68) described the use of RASSF1A for monitoring tumor shrinkage after neoadjuvant chemotherapy by collecting consecutive serum samples from 52 patients with locally advanced breast cancer. All patients had monthly physical examinations during therapy and tumor assessment by mammography and breast ultrasound before the start of chemotherapy and again after therapy but before surgery. Based on the clinical examination and imaging studies, patients were classified as complete responders, partial responders and minimal or non-responders. Whole blood was collected before each of 7 consecutive neoadjuvant chemotherapy cycles (Adriamycin Cytoxan or Taxol trastuzumab) and analyzed for RASSF1A. The cutoff value was set to 1%. RASSF1A hypermethylation was detected in serum of 21 patients before therapy. RASSF1A methylation became undetectable in serum early during therapy in 4 patients who had complete response. In contrast, serum RASSF1A persisted longer or throughout the treatment in 17 patients who had partial or minimal pathological response.
Zurita et al (52) investigated the clinical usefulness of ESR1 and SNF as biomarkers for treatment efficacy in metastatic breast cancer patients. Blood samples were obtained from 34 consecutive patients on the day that each chemotherapy cycle was started, giving a series of sequential samples. The ratio of the SNF level before the first chemotherapy cycle to the level just before administration of the second chemotheraphy cycle was calculated. This was done for 2 groups of patients displaying “continuous decline” and for patients displaying “rise-and-fall” in SNF levels, and was used for calculating a cutoff level for the ratio of the SNF. The correlation between SNF methylation and breast cancer metastasis and progression suggests an application of SNF methylation as a biomarker to monitor adjuvant treatment response (52). These findings (52, 67, 68) support further development of assays for monitoring response during therapy.
Unfortunately, monitoring studies are difficult to handle and no consensus yet exists on the optimal design and conduct of longitudinal trials involving serial monitoring of tumor biomarkers (69). One proposal is a 4-phase model for monitoring trials analogous to that in use for the investigation of new drugs (69). Initially, phase I biomarker trials should examine the relationship between a change in tumor burden and a change in serial biomarker concentrations in non-blinded investigations. To do so blood sampling must be properly timed after the chemotherapy cycles since DNA-methylation levels may increase transiently after an effective chemotherapy treatment and produce false-positive results. Provided that an observed change in the concentrations of DNA-methylation markers exceeds estimates of total variation (biological and analytical), the change is likely to reflect a modification in disease activity. The reference change value, i.e. the change between 2 results that must be exceeded before the change can be considered clinically significant, provides a convenient means of assessing whether the measured change is caused by altered disease activity (70, 71). If altered disease activity is observed, the acute change in biomarker must have influence on clinical-decision making, e.g. additional diagnostic tests, imaging or a change in therapy. Last, the most important question to address is whether introducing monitoring with the new biomarker has a beneficial effect on outcome, such as overall survival (69).
Discussion
Aberrant DNA methylation has some analytical features that are appealing and potentially useful. First, DNA molecules are relatively stable even in water solutions stored at room temperature. Isolated DNA can be stored almost indefinitely at −80°C. Second, DNA can be obtained from different sources, including serum (7-9), blood (12, 13), nipple aspirate, and lavage fluid (3, 20). Serum and plasma are more readily accessible body fluids and the collection of a sample is patient-friendly and does not require a specialist. It appears that serum contains more DNA than plasma does. However, the serum DNA concentration may not necessarily imply a relative higher concentration of tumor-derived DNA. There is even some evidence that DNA is released from the tumor as a glyconucleoprotein complex that may protect it from degradation (6). Most importantly, the discovery that DNA can be shed into the bloodstream in detectable DNA concentrations opens for validation of a diagnostic, prognostic, monitoring and follow-up tumor marker for breast cancer. However, no studies have been conducted to determine if non-invasive sampling from the breast (aspirates) gives an analytical advantage compared with blood sampling.
The greatest potential for improving life expectancy may be in early detection and screening. Few studies have investigated the potential of DNA methylation for initial screening of large cohorts of healthy women, and monitoring studies of the tumor burden during chemotherapy before (neoadjuvant) and after (adjuvant) surgery. There is a lack of consensus on the choice of analytical methods and how to determine cutoff values when serial changes in concentrations are required, e.g. in monitoring studies. Finally, it remains to be seen whether modifications in clinical testing strategies based on the analysis of DNA methylation will improve overall survival rates.
In conclusion, many studies have investigated the potential of DNA-methylation markers for clinical diagnosis and prognosis, as shown in Table I. This survey ranks RASSF1A as the biomarker with the highest clinical sensitivity (56%). SNF and HIN-1 were found to have a higher clinical sensitivity (74% and 60%, respectively), but data could only be calculated from 4 independent studies for SNF (14, 36, 67, 72) and 3 studies for HIN-1 (32, 36, 51). Given the large variation in sensitivity between studies for the same biomarker, more studies are needed to validate SNF and HIN-1 as superior to RASSF1A. Promising robust candidates may then include RASSF1A, CDH1, CCND2, ESR1 and APC. The clinical specificity was found to be as high as nearly 100% for RASSF1A (7, 8, 15, 73, 74). Overall, a panel of biomarkers increases the clinical sensitivity. A panel with 3-5 markers may encompass all types of breast cancer, stage, grade, receptor status, degree of invasion, etc. Discovery of more biomarker genes will further improve the analytical sensitivity. Regardless of this, development and validation of one standard analytical method will be required before the method can be evaluated in multiple clinical trials against standard care.
Abbreviations
estrogen receptor (ER)
human epidermal growth factor receptor 2
luminometric methylation assay
multidrug resistance protein 1
long interspersed nuclear repetitive elements
progesterone receptor
Gene symbols
Adenomatous polyposis coli (APC); ADP-ribosylarginine hydrolase (ARH1); ataxia telangiectasia mutated (ATM); ATP-binding cassette B1 (ABCB1); breast cancer 1 and 2 (BRCA1 and BRCA2); catenin beta 1 (CTNNB1); cyclin D2 (CCND2); cyclin-dependent kinase inhibitor 2A (CDKN2A); cyclin-dependent kinase 10 (CDK10); cysteine dioxygenase type 1 (CDO1); cytochrome P450 1B (CYPB1); cytokine high in normal type 1 tumor suppressor (HIN-1); death-associated protein kinase 1 (DAPK1); E-cadherin (CDH1); estrogen receptor α (ESR1); forkhead box C1 (FOXC1); glutathione S-transferase pi (GSTP1); H-cadherin (CDH13); methylguanosine-DNA-methyltransferase (MGMT); nestin 1 (NES1); paired-like homeodomain transcription factor 2 (PITX2); phosphoserine aminotransferase (PSAT1); rat sarcoma associating domain family 1A (RASSF1A); retinoic acid receptor β gene (RARB); reversion induced LIM (RIL); stratifin (SNF); target of methylation-induced silencing (TMS1); thrombospondin 1 (THBS1); tissue inhibitor of metalloprotease 3 (TIMP3); twist-related protein 1 (TWIST1).