
Abstract
Upon transmission, human immunodeficiency virus type 1 (HIV-1) establishes infection of the lymphatic reservoir, leading to profound depletion of the memory CD4+ T cell population despite the induction of the adaptive immune response. The rapid evolution and association of viral variants having distinct characteristics during different stages of infection, the level of viral burden, and rate of disease progression suggest a role for viral variants in this process. Here, we review the literature on HIV-1 variants and disease and discuss the importance of viral fitness for transmission and disease.
Transmission and Selection of Variants
Transmission of HIV-1 can occur via sexual, parenteral, or vertical routes of infection (Lamers et al. 1993; Mulder-Kampinga et al. 1993; Pang et al. 1992; Scarlatti et al. 1993; Wolfs et al. 1992; Wolinsky et al. 1992; Zhang et al. 1993; Zhu et al. 1993). Each of these represents a distinct environment and therefore a distinct set of factors affecting selection of viral variants. Research focusing on factors affecting HIV-1 selection has addressed a multitude of issues ranging from stochastic versus selective models of transmission, single versus multiple variant transmissions, cell-free versus cell-associated virus transmission, selection criteria for restricting variant transmission and compartmentalization of variants within hosts biasing variant selection during transmission.
In the context of sexual transmission, the type of sexual encounter, gender of the transmitter and recipient, nature of the mucosal surface and presence of other genital tract infections can all play an integral role in selection during transmission. In order to distinguish the presence or absence of viral variants, the highly variable sequences of HIV-1 envelope (env) have been used as markers of diversity, especially in the V1–V2 and V3 regions of env which are important for immune recognition (Clerici et al. 1991; Javaherian et al. 1990; LaRosa et al. 1991), replication efficiency (Takeuchi et al. 1991) and cellular tropism (Chesebro et al. 1991; de Jong et al. 1992; Hwang et al. 1991; O'Brien et al. 1990; Shioda et al. 1991; Westervelt et al. 1991; Westervelt et al. 1992) and are under constant selection, resulting in a high rate of variation (Delwart et al. 1994; Delwart et al. 1993; Holmes et al. 1992; Javaherian et al. 1990; Kuiken et al. 1993; Wang et al. 1995; Zhu et al. 1993). Due to the increased variability, these regions have been used extensively to characterize the level of heterogeneity in donor/recipient HIV-1 transmission events. Transmission of variants found in low abundance (minor variants) would argue against a stochastic transmission model and many reports find that minor variants are often the predominant variants in the newly infected host (Zhu et al. 1996; Zhu et al. 1993; Wolfs et al. 1992; Wolinsky et al. 1992; Zhang et al. 1993). The majority of reports find that HIV-1 env sequences in newly infected individuals are relatively homogenous, despite the fact that the transmitters harbor heterogeneous genotypes (Delwart et al. 2002; Derdeyn et al. 2004; Learn et al. 2002; Wolinsky et al. 1992; Wolfs et al. 1992; Zhang et al. 1993; Zhu et al. 1993; Zhu et al. 1996). This sequence homogenization relative to the sequence heterogeneity of the transmitter could be a result of selection based on low inoculum levels (i.e. a founder effect), selective penetration of virus from donor to recipient and/or selective amplification of particular viral variants within the newly infected recipient. Factors such as cell-free or cell-associated virion transmission are also thought to affect virus transmission (Ludlam et al. 1985; Zhu et al. 1996). For instance, cell-free virions could be more likely to result in one or a few viral variants being transmitted, whereas cell-associated transmission could increase the likelihood of a multiple variant transmission event. Recent data support the complexity of transmission events in that some infections were initiated from a single virus while other infections resulted from a multi-virus transmission event (Keele et al. 2008). Gender differences also play a role in determining variant transmission profiles (Grobler et al. 2004; Haase, 2005; Long et al. 2000; Long et al. 2002; Ritola et al. 2004; Poss et al. 1995), potentially due to the type and duration of exposure to mucosal surfaces which may favor single or multiple viral variant transfers (Pilcher et al. 2004a; Pilcher et al. 2004b; Pope and Haase, 2003; Sagar et al. 2004; Vernazza et al. 1999; Wawer et al. 2005). It remains to be determined whether these differences are based on mucosal tissue morphology or on more complex biological factors.
Vertical transmission or mother-to child transmission (MTCT) has also been extensively studied and research has attempted to address similar questions as with sexual transmission. However, with MTCT, an additional layer of factors regarding the temporal nature of the transmission event arises. Specifically, does transmission occur prepartum (in utero), intrapartum (at delivery) and/or post-partum (via breast feeding) and what, if any, effect does this have on variant transmission profiles? Studies have found that transmission of virus can occur during any of these phases of pregnancy, but the effect on variant transmission remains elusive (Courgnaud et al. 1991; Ehrnst et al. 1991; De Rossi et al. 1992; Lepage et al. 1987; Soeiro et al. 1992; Ziegler et al. 1985). Similar to sexual transmission, major, minor and multiple variant transmission events are also seen in MTCT cases (Dickover et al. 2001; Kliks et al. 1994; Lamers et al. 1994; Narwa et al. 1996; Nowak et al. 2002; Briant et al. 1995; Pasquier et al. 1998; Wade et al. 1998; van't Wout et al. 1994). However, most studies have found single variant transmission events and greater homogeneity within the newly infected child when compared to the mother, suggesting selective pressures during transmission (Dickover et al. 2001; Becquart et al. 2002; Nowak et al. 2002; Wike et al. 1992; Wolinsky et al. 1992; Mulder-Kampinga et al. 1993; Ahmad et al. 1995; Roth et al. 1996; Scarlatti et al. 1993). Whether these selective criteria are the same or similar to those involved in sexual transmission remains unknown.
Quasi-Species Evolution Post Transmission
Once a successful transmission event has occurred, mutations in the viral genome, host immune pressures and target cell availability drive HIV-1 diversification and evolution, eventually resulting in viral variants that differ from the founding parent viruses. While there may be a slight loss in replicative capacity initially, these new variants are capable of evading host immune defenses, persisting, and eventually driving CD4+ T cell depletion and progression to AIDS.
HIV-1 evolution is rapid due to a high viral replication rate, an error-prone replication of reverse transcriptase (Coffin, 1995; Mansky and Temin, 1995; Preston et al. 1988; Roberts et al. 1988), transcription by host RNA polymerase II (Laakso and Sutton, 2006), recombination events between co-infecting HIV-1 viral variants (Charpentier et al. 2006; Coffin, 1995; Jung et al. 2002; Kemal et al. 2003; Levy et al. 2004; Philpott et al. 2005; van Rij et al. 2003; Zhuang et al. 2002) and rapid immune system-mediated selection of viral variants (Jung et al. 2002; Williamson et al. 2005). The combination of these events continually drives HIV-1 diversity in infected hosts (Fig. 1). However constraints are placed on the mutations that can be incorporated into the viral genome due to structural and functional requirements of the encoded proteins involved in viral replication (Draenert et al. 2006).
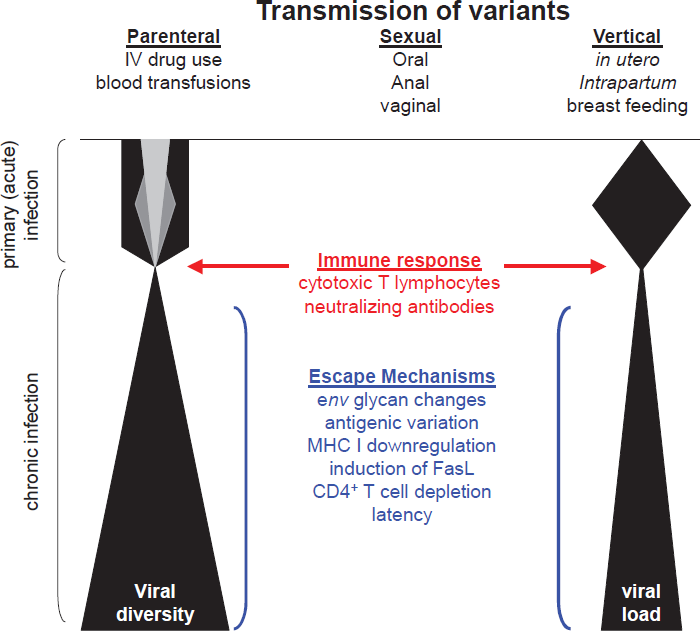
Schematic diagram representing changes in viral diversity and viral load throughout the course of infection. The hypothetical amounts of viral diversity at or shortly following transmission are indicated by the different shadings during primary infection. Factors that may influence viral diversity, selection, and persistence are shown.
During the initial stages of infection, selected mutations appear to favor immune escape rather than enhanced viral replication (Martinez-Picado et al. 2006; Leslie et al. 2004; Goepfert et al. 2008). However, late during the asymptomatic chronic stage of infection, when immune pressures have been ablated by destruction of immune cells or exhaustion of immune responses, mutations increasing viral replication begin appearing or reappearing within the virus population (Mild et al. 2007). These data suggest that early during infection mutations are selected based on immune evasion rather than enhanced viral replication, whereas during later stages of infection more pathogenic, but less immune-evasive viruses appear to drive disease progression. This hypothesis is supported by studies of SIV in macaques, where there is a fitness cost to escape from CTL responses early after infection (Friedrich et al. 2004). However, with time, variants having increased replicative capacity emerge and eventually drive disease progression (Kimata et al. 1999; Rudensey et al. 1995).
While most basic and clinical studies seem to agree that variants isolated early during infection appear to be less pathogenic and have lower replicative capacity compared to late stage isolates, mathematical modeling of within-host virus evolution suggests that over the course of host infection viral variants move toward reduced replicative fitness (Wodarz and Levy, 2007; Ball et al. 2007). This apparent discrepancy could result from the fact that mathematical modeling has not accounted for the plethora of complex factors that play roles in viral fitness in vivo. However, one would be remiss to disregard key aspects of these mathematical models. Indeed, the overall conclusion from mathematical modeling is that the most replicatively fit HIV-1 viral variant would not be able to sustain infection, especially as is seen in chronic HIV-1 infections, due to the rapid destruction and depletion of the essential target CD4+ T cell population. Thus, an evolutionary ceiling is placed above viral replicative fitness. By contrast, studies examining replicative fitness do indicate that as infection progresses more pathogenic viral variants with higher replicative capacities appear at late stages of infection (Kimata et al. 1999; Kimata, 2006). Interestingly, both the mathematical models and clinical studies demonstrate that, even in the presence of these pathogenic and replicatively fit viral variants, less pathogenic and replicatively robust viral variants are still found in circulation (Ball et al. 2007; Wodarz and Levy, 2007; Mild et al. 2007; Mansky and Temin, 1995; Gali et al. 2007).
Additional correlates of AIDS disease progression include slower rate of synonymous substitution rates, indicative of general, non-selective mutation rates, (Lemey et al. 2007; Stilianakis and Schenzle, 2006), increased viral replication (Kimata et al. 1999; Birch et al. 2001; Dyer et al. 1999; Kirchhoff et al. 1995; Learmont et al. 1999), persistent immune activation (Bofill et al. 1996; Grossman et al. 2006; Giorgi et al. 1999; Sousa et al. 2002), broad-range CTL responses (Karlsson et al. 2007; Fernandez et al. 2007) and specific host human leukocyte antigen (HLA) class I alleles (Carrington et al. 1999; Trachtenberg et al. 2003).
Correlates of Pathogenicity: Phenotypic Changes during Viral Infection
Phenotypic characteristics of HIV-1 that have been extensively studied for correlation to disease progression include replicative capacity (also commonly referred to as replicative fitness), which is generally classified as rapid/high or slow/low in relation to replication and production of virus (De Rossi et al. 2005; De Rossi et al. 1993; Connor et al. 1993), syncytium induction, classified as non-syncytium inducing (NSI) or syncytium inducing (SI) virus (Koot et al. 1992; Jurriaans et al. 1994), co-receptor usage with the vast majority of viral variants being classified as CCR5-using (R5), CXCR4-using (X4) or dual-tropic (R5X4) viruses (Littman, 1998; Doms and Peiper, 1997) and macrophage-tropic (M-tropic) or T cell-tropic (T-tropic) variants. Previously, it was believed that these phenotypic characteristics were intimately linked such that rapid/high viral variants were also SI, X4, T-tropic variants and slow/low viruses were NSI, R5, M-tropic variants (Alkhatib et al. 1996a; Tersmette et al. 1988; Tersmette et al. 1989). However, while there is a correlation with co-receptor usage, SI ability and tropism, these are separable phenotypic features and therefore should be individually tested for when characterizing viral isolates (Aquino-de Jesus et al. 2000; Peters et al. 2006).
Following transmission, R5-tropic viruses typically predominate early stages of infections (Connor et al. 1997). However, it remains unclear as to whether R5 viruses are the only viruses transmitted or whether both R5 and X4 viruses can be transmitted but that X4 viruses are less fit, resulting in only R5 variants being detected during the early stages of infection. These virus isolation studies also show that X4 and dual-tropic viruses generally are not detected until very late in infections at the juncture of transition from asymptomatic infection to AIDS. Morever, X4 variants are only found in 50% of cases (Berger et al. 1999), demonstrating that X4 variants are not required to drive progression to AIDS (Campbell et al. 2003; Kimata et al. 1999; Kwa et al. 2003; Koot et al. 1993; Tersmette et al. 1989).
There is experimental evidence that X4 variants may be more susceptible to control by CD8+ cytotoxic T cells than R5 viruses (Harouse et al. 2003). Indeed, X4 variants may be rapidly selected against during primary infection, allowing R5 variants to emerge and predominate in the infection. Thus, X4 viruses appearing late in infection may not necessarily drive disease progression, but rather serve as indicators of an exhausted and dysfunctional immune system, which allows an unchecked replication of viruses and destruction of the remaining CD4+ T cell population.
Cell tropism has also been extensively studied in vivo and in vitro. A current model is that M-tropic viruses predominate during early stages of transmission, since it is believed that tissue-resident macrophages, monocytes and dendritic cells are initial cell targets during the actual transmission event. While infection of these cell types remain important throughout the course of disease progression in regard to latently infected cell populations (Aquino-de Jesus et al. 2000), a shift in cell tropism occurs early and rapidly as virus is trafficked from the site of infection to lymphatic tissues where robust replication can take place in CD4+ T cells.
Lastly, there appears to be selection of NSI variants during transmission (van't Wout et al. 1994; Zhu et al. 1993; Tersmette et al. 1988; Keet et al. 1993), but whether the phenotypic change from the transmitted NSI virus to a SI variant is important for progression to AIDS requires further exploration. Virological studies indicate that the switch from NSI to SI phenotype is not required for AIDS progression (Fitzgibbon et al. 1998; Spencer et al. 1994), but may increase the rate of AIDS progression (Fauci, 1996; Glushakova et al. 1998).
A simple explanation of the appearance of each of these phenotypic characteristics during infection is that as viral diversity increases the rate of AIDS progression increases, thus the diversity, as indicated by the appearance of these phenotypes, rather than the functions of the respective phenotypes drives AIDS progression (Sagar et al. 2003). Therefore it may not be the presence per se of X4 and SI viral variants, but rather the weakening of selective pressures from the host immune response on generalized viral replication, allowing an outgrowth of previously immune response-targeted phenotypes which serve simply as indicators of the weakened and dysfunctional immune response (Troyer et al. 2005).
Finally, changes in N-linked glycosylation and length of Env variable regions V1 and V2 have been reported to occur with infection. Initially, it was observed in SIV-infected macaques that variants with limited N-linked glycans in the V1/V2 region of Env dominated the early stages of infection (Overbaugh and Rudensey, 1992). As the animals progressed to disease, additional N-linked glycosylation sites appeared and the V1/V2 region lengthened. Some of these changes correlated with protection from neutralizing antibodies and loss of macrophage tropism (Rudensey et al. 1995; Rudensey et al. 1998). More recently several groups have reported similar glycosylation changes to occur with HIV-1 (Chohan et al. 2005; Derdeyn et al. 2004; Sagar et al. 2006; Wu et al. 2006) following sexual transmission in discordant couples and vertically from mother to child. However, while less glycosylated Env proteins from early stages of infection were associated with sensitivity to neutralizing antibodies after sexual transmission, this was not observed with vertically transmitted mother to child variants. These data raise questions about the functional changes in Env conferred by additional glycans. Recent studies demonstrate that GAG-specific CTL responses have a more profound impact on pathogenicity and viral load than do the ENV-specific CTL response (Kiepiela et al. 2007; Peut and Kent, 2007), lending support to the hypothesis that env-mediated phenotypic changes serve as indicators of dampened immune selection rather than functional mediators of pathogenicity.
Viral Determinants Altering Phenotype
Phenotypic differences among viral variants have been studied and mapped to specific regions of the viral genome, including env, pol and nef. The env gene is a major determinant in viral replicative fitness as its protein products, gp120 and gp41, mediate cell binding via the receptor and co-receptors and fusion of the cellular plasma membrane and the viral membranous envelope (Baribaud and Doms, 2001; Berger et al. 1999; Poignard et al. 2001). Numerous studies have documented that env sequences influence viral transmission (Hsu et al. 2003; Tersmette et al. 1988), cell tropism (Berger, 1997; Hoffman and Doms, 1999; Alkhatib et al. 1996b; Choe et al. 1996; Deng et al. 1996) and are major targets of the host immune response (Levy, 1993; Richman et al. 2003; Wei et al. 2003b), including both CTL and neutralizing antibody responses (Jones et al. 2004; Borrow et al. 1997; Geels et al. 2003). Furthermore, env sequences appear to have the greatest impact on competitive viral replicative fitness in vitro in comparison to other regions of the viral genome (Ball et al. 2003).
The phenotypic change in co-receptor usage by viral variants has been mapped to env, specifically the V1/V2 and V3 regions. Mutations in V3 have been extensively studied and found to directly control tropism usage (Cocchi et al. 1996; Harrowe and Cheng-Mayer, 1995). However, mutations responsible for the switch from CCR5- to CXCR4-usage appear to confer a replication fitness disadvantage to the resulting virus (Wagner et al. 1999; Kelleher et al. 2001), either due to decreases in evasion of host CTL responses, enhanced sensitivity to antiviral drugs or decreased avidity for receptor/co-receptor molecules (Marozsan et al. 2005; Lobritz et al. 2007; Derdeyn et al. 2000; Labrosse et al. 2003; Strizki et al. 2001; Torre et al. 2000; Trkola et al. 1998). Mutations in the V1/V2 region also play a role in co-receptor usage (Groenink et al. 1993; Koito et al. 1994; Koito et al. 1995; Ogert et al. 2001; Ross and Cullen, 1998; Sullivan et al. 1993; Toohey et al. 1995; Wyatt et al. 1995; Yoshimura et al. 1996) with an apparent ability to compensate for the loss-of-fitness mutations in the V3 region, thus allowing the co-receptor switch to occur (Pastore et al. 2006). It is interesting to note that relatively few mutations are required for the R5/X4 co-receptor switch, but that this phenotypic change does not occur for several years after initial infection (Schuitemaker et al. 1992; Shankarappa et al. 1999). It is hypothesized that this delay in emergence of X4 variants is in part due to a very limited number of viable mutational pathways of transitional viral variants that maintain a minimally competitive replication fitness and evasion from host CTL responses (Pastore et al. 2006; Fernandez et al. 2005; Peyerl et al. 2004). Env mutations are also responsible for the NSI/SI phenotypic changes. Mutations responsible for this phenotypic switch have not been studied as extensively; however, due to the high degree of correlation between NSI/SI and R5/X4 phenotypic switching it is likely that mutations reside in the same sequences of env (e.g. V1/V2 and V3).
Functional reverse transcriptase (RT) is a heterodimer composed of subunits, p66 and p51, with p66 containing active RT and RNase H activity (Veronese et al. 1986). The majority of RT mutations and variants that affect functionality have been identified during studies with antiviral drugs designed to target RT-specific catalytic steps in the viral lifecycle. These drugs include nucleoside reverse transcriptase inhibitors (NRTIs) and nonnucleoside reverse transcriptase inhibitors (NNRTIs) that inhibit RT activity by acting as DNA chain terminators or steric inhibitors of nucleoside binding, respectively. While it is clear that during highly active antiretroviral therapy (HAART) specific mutations are selected within RT that decrease sensitivity to prescribed drug regimens (Gu et al. 1992; Gao et al. 1993; Tisdale et al. 1997; Walter et al. 2002), some of these mutations can have positive effects on virus replication and fitness (Hu et al. 2007) while others have deleterious effects on replication (Wakefield et al. 1992; Larder et al. 1995; Olivares et al. 2004; White et al. 2002). The majority of these mutations appear to affect RT affinity for dNTPs which in turn alter RT DNA polymerase fidelity and/or processivity (White et al. 2002; Jonckheere et al. 1998; Bebenek et al. 1995; Back et al. 1996; Wainberg et al. 1996; Hsu et al. 1997; Feng and Anderson, 1999). Since deleterious mutations are more frequent than advantageous mutations within any genome, extreme RT infidelity would quickly result in non-functional HIV-1 genomes (Furió et al. 2005; Kimura, 1967). Conversely, increased RT fidelity would impact the intrinsic ability of HIV-1 to evade the host immune response and escape from HAART, thus RT fidelity is an intricate balance of genomic diversity and stability (Furió et al. 2007). Several studies have identified available dNTP levels as a factor that can affect RT fidelity (Back and Berkhout, 1997; Vartanian et al. 1997), yet very little research has been performed with HIV-1 RT fidelity mutations that occur naturally within infected hosts in the absence of HAART, despite the fact that cell types known to be targets for HIV-1 infection (e.g. macrophages, resting T cells, activated T cells) have variable dNTP pools (Traut, 1994; Diamond et al. 2004; Hauschka, 1973; Fuller et al. 1982; Skoog and Bjursell, 1974; Yao et al. 2003). A recent study with SIV RT variants isolated from infected macaques demonstrated rapid selection against an RT variant with higher replication fidelity (Biesinger et al. 2008). These data suggest that RT's ability to misincorporate dNTPs in settings with limited resource availability or the indirect effect misincorporation has on processivity allows for more rapid genomic reverse transcription and integration and is an important aspect of replication fitness. Regardless, it is clear that intrinsic RT properties (e.g. fidelity and processivity) are important factors in determining viral replicative fitness in the context of host cell usage and overall viral production.
Nef is a multifunctional viral protein with key roles in the viral life cycle which include down-regulation of CD4, CD28 and class I major histocompatibility complex (MHC-I), alteration of CD4+ T cell activation, enhancement of viral infectivity and replication and sensitization to apoptotic pathways (Laforge et al. 2007; Anderson and Hope, 2004; Collins and Baltimore, 1999; Johnson and Desrosiers, 2002; Renkema and Saksela, 2000; Roeth and Collins, 2006; Wei et al. 2003a). The essential nature of Nef activity for viral replication in vivo has been well-documented. However, the details of how Nef is able to exert its pleiotropic effects remain poorly described. Nef functionality in vivo exerts a dramatic effect on viral fitness and progression to AIDS. However, which function of Nef provides this enhancement is still unclear. Several possibilities, although not mutually exclusive, have been presented in the literature, including CTL escape via MHC-I downregulation (Ali et al. 2003), modulation of T cell activation via interactions with cellular factors such as PAK2 (Lu et al. 1996) and increased host cell apoptosis through unknown cellular interactions (Laforge et al. 2007). Finally, SIV-macaque studies suggest that the major function of Nef may be to enhance virion infectivity, as nef mutants that fail to enhance viral infectivity replicated poorly in vivo (Iafrate et al. 2000; Patel et al. 2002). Despite the fact that nef is essential for robust HIV-1 replication in vivo and evasion from the host immune response, it has recently been reported that highly attenuated nef-deleted viral infections still resulted in HIV-associated diseases and loss of CD4+ T cell levels (Gorry et al. 2007), suggesting that Nef functions are dispensable in context of AIDS progression over extended periods of time.
Model Systems to Measure Viral Phenotype and Fitness
Correlations of viral phenotype and replicative fitness are found throughout the literature. However, most studies to determine phenotype and phenotypic changes (e.g. tropism, infectivity and ability to induce syncytia) are generally performed in in vitro single virus replication assays. The classical definition of fitness has necessarily required some aspect of direct competition for limited resources; therefore single-virus infections and assays are unable to be applied to directly answer whether one viral variant is more fit in a given environment versus another variant in the same environment. Relatively few studies have combined studies of viral phenotype with dual-virus replication assays to determine viral variant fitness.
In vivo studies to determine fitness of HIV-1 in the presence of immune responses have been hampered by the lack of an animal model to validate observations, and collection of samples from co-infections or super-infections within human cohorts are limited (Piantadosi et al. 2007; Kozaczynska et al. 2007; Gottlieb et al. 2007). In vitro competition assays have been used to characterize general viral fitness in regards to infectivity, replication and cytopathicity. These types of experiments suggest increasing fitness trends in the overall HIV-1 population over time despite the ‘reset’ of viral phenotype that occurs during each transmission event (Gali et al. 2007). Additionally, competition assays demonstrated that R5 and X4 phenotype do not necessarily correlate with lower or higher replicative fitness, respectively. One study found that R5 and X4 clones were equally fit in terms of viral replication in mitogen-activated T cells, but that X4 viruses did appear to have a selective advantage in DC-T cell co-cultures (Arien et al. 2006), while another study found that some R5 clones were more fit than X4 clones (Quinones-Mateu et al. 2000). These observations could help explain why the shift from R5 to X4 is not essential for progression to AIDS, since mass activation of the host CD4+ T cell population is characteristic of late stages of HIV-1 infection allowing outgrowth of either R5 or X4 viruses, but that moderate increases in viral spread which is correlated with X4 clones, would shorten the time to AIDS onset. Specific mutations in env have now been correlated with changes in fitness and entry inhibitor resistance (Lobritz et al. 2007; Rangel et al. 2003). Competition assays have also been used to explore the effects of environmental factors on viral fitness, such as the availability of permissive cell populations (Goodenow et al. 2003). Finally, the analysis of HIV-1 superinfection in infected individuals provides a rare glimpse into viral fitness in vivo (Gottlieb et al. 2007; Blish et al. 2007; Kozaczynska et al. 2007). However, research is limited by the rarity of these occurrences and knowledge of the properties of the viruses.
Because of the limitations for studying HIV-1 fitness in vivo, infection of macaques with SIV has remained a critical model for exploring questions about HIV-1 fitness and pathogenesis. The model system allows incorporation of viral fitness scores in the presence of shifting target cell populations, immune response and host variability and in context of infection with a related lentivirus. It also enables fitness to be examined with variants of known in vitro phenotype and in vivo pathogenicity. Indeed, a recent study showed that a SIV variant with increased replicative capacity and pathogenicity demonstrated higher competitive viral replication fitness in vitro compared to the slower-replicating and minimally pathogenic parental clone (Voronin et al. 2005). These types of studies provide important experimental evidence in support of the hypothesis that viral fitness impacts HIV-1 pathogenesis.
The macaque model system has also been used to study effects of dual virus infections in the host. These studies have mainly used R5- and X4-tropic SHIV chimeric constructs (Burke et al. 2006; Wolinsky et al. 2004; Otten et al. 1999). These dual virus infection studies are vital to understanding viral fitness in the full context of an infection where both viral and host factors are present and influence fitness outcomes of variants. However, few studies have been performed using dual-virus infections to specifically study the relative fitness of variants and which determinants are able to confer fitness advantages at various stages of infection and disease states (Harouse et al. 2003). Furthermore, these studies have been limited to competitions with R5-tropic and X4-tropic viruses. Additional studies with different variants, including R5-tropic viruses with distinct phenotypes, would further enhance our understanding of viral determinants influencing transmission, persistence and disease.
Conclusion
Route of HIV-1 transmission appears to dictate to some extent the type and number of viral variants that comprise the founder population of viruses in a newly infected host. Particularly interesting, as discussed above, are the aspects controlling selection during transmission where different factors, ranging from time of contact between viruses trapped within the vaginal cavity versus rectal tissues to cell-free virions versus cell-associated viruses to the glycosylation patterns found on gp120, appear to affect the number and type of transmitted viruses. Additionally, host immune responses and the corresponding counter-defenses of the infecting variants that enable evasion of the immune response represent an environment of attrition. While the strength of the immune response may initially determine viral set-points and rates of CD4+ T cell loss, latency and high mutability enable HIV-1 to subvert some host immune responses and evade other immune responses, leading to profound depletion of CD4+ T cell populations and onset of AIDS.
Correlating viral phenotypes with pathogenicity has resulted in identification of regions of interest within the viral genome that impact replication and persistence. Added studies of competitive replication fitness will be necessary to shed more light on the significance of these changes for viral fitness at early and late stages of infection. In this regard, the infection of macaques with SIV variants will be needed to provide invaluable results for deciphering fitness determinants for transmission and pathogenesis.
Footnotes
Acknowledgments
J.T.K. is supported by NIH grant R01 AI047725 and T.B. is supported by an NIH training grant in Molecular Virology (T32-AI07471).